A lot of systems involve DNA or RNA. Cpptraj can be used to analyze the backbone structure in a variety of ways. Helpful lessons on nucleic backbone analysis be found on the Case Group page and on the website for the 3DNA program.
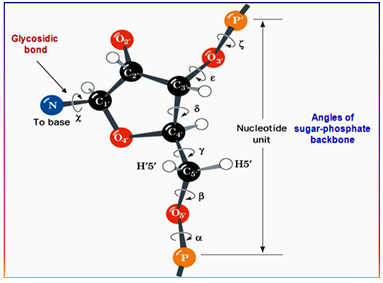
The different backbone angles can be seen in the figure above. These backbone angles have been defined through:
#Alpha= :x-1@O3' :x@P :x@O5' :x@C5'
#Beta= :x@P :x@O5' :x@C5' :x@C4'
#Gamma= :x@O5' :x@C5' :x@C4' :x@C3'
#Delta= :x@C5' :x@C4' :x@C3' :x@O3'
#Epsilon= :x@C4' :x@C3' :x@O3' :x+1@P
#Zeta= :x@C3' :x@O3' :x+1@P :x+1@O5'
## Chi examples
## Pyrimidines (Y)= :x@O4' :x@C1' :x@N1 :x@C2 [C, T, U]
## Purines (R)= :x@O4' :x@C1' :x@N9 :x@C4 [A, G]
Thus, using the dihedral command in cpptraj can give you information on
these angles.
Specifying a dataset name (ex. alpha449) will allow multiple angles to be
printed to the same out file.
#Residue 449 DG
dihedral alpha449 :448@O3' :449@P :449@O5' :449@C5' out RNA_backbone-449-dihed.dat
dihedral beta449 :449@P :449@O5' :449@C5' :449@C4' out RNA_backbone-449-dihed.dat
dihedral gamma449 :449@O5' :449@C5' :449@C4' :449@C3' out RNA_backbone-449-dihed.dat
dihedral delta449 :449@C5' :449@C4' :449@C3' :449@O3' out RNA_backbone-449-dihed.dat
dihedral epsilon449 :449@C4' :449@C3' :449@O3' :450@P out RNA_backbone-449-dihed.dat
dihedral zeta449 :449@C3' :449@O3' :450@P :450@O5' out RNA_backbone-449-dihed.dat
dihedral chi449 :449@O4' :449@C1' :449@N9 :449@C4 out RNA_backbone-449-dihed.dat
Double-stranded nucleic acids can also be studied with respect to other base
pairs.
Information on different base pairs can be gathered by using the nastruct
command in cpptraj.
This command will automatically determine what is paired together, and
nonstandard residues can be calculated based on the original base it was
derived from (using resmap.
The nastruct command has predetermined prefixes (BP.; BPstep.; and
Helix.), but you specify the rest of the filename and extension after naout.
An example is shown below.
nastruct master resrange 431,432,433,434,435,446,447,448,449,450 naout master.dat \
resmap 5xC:C calcnohb
Because nastruct will automatically match pairs, the output will need to be
cleaned up before plotting any data.
This can be done pretty easily using awk.
$ awk 'NR == 1 || NR % 5 == 2' BP.master.dat > BP-A2-T2.dat
$ awk 'NR == 1 || NR % 4 == 2' BPStep.master.dat > BPstep-A2-C3.dat
$ awk 'NR == 1 || NR % 4 == 2' Helix.master.dat > Helix-A2-C3.dat
The NR == 1 will print the first row (the header) and the NR % 4 == 2 will
print every 4th row starting with the second row.
Using 3DNA
Another program that can be used to analyze DNA is 3DNA program. The program can be downloaded after making an account on the 3DNA forum. A downside to using 3DNA is that you cannot analyze your entire trajectory–only snapshots in the form of PDBs. That said, the information is printed very cleanly, and the analysis takes seconds.
After installation and selecting your snapshots, the following command will be used to gather information on form, sugar puckering, and more.
$ find_pair WT_protein_system_md50.pdb | analyze
3DNA also has 2 other programs in testing, SNAP and DSSR.
Help for any 3DNA analysis can be acquired by doing the executable name with
the -h flag.