CAVER is a software tool for analyzing tunnels in biomolecular structures.
There are several tools: (a) CAVER, a command-line tool; (b) CAVER Analyst, a GUI; and (c) a CAVER PyMOL plugin.
CAVER (Command-Line)
The command-line version requires an input file, described on the
next page. Once the input file is generated,
you can run it using a script, like run-cluster.sh below.
#!/bin/bash
## Set program location (can be in .bashrc instead)
export CAVER="/home/$USER/bin/caver_3.0/caver"
## Areas of red conservation
##
## 601 is metal center
## 603 is cofactor
listVar=(601 603)
## Backup your original config file
cp config.txt config-start.txt
for RESNUM in "${listVar[@]}"
do
## Use double quotes so the shell can substitute variables
## Replace the full line with "starting_point_residue $RESNUM"
sed "22c\starting_point_residue $RESNUM" config-start.txt > config.txt
## Run the Caver program
java -Xmx10000m -cp $CAVER/lib -jar $CAVER/caver.jar -home $CAVER -pdb ./ -conf ./config.txt -out ./caver_output_res$RESNUM
## Copy the config file used to the output folder
cp config.txt ./caver_output_res$RESNUM/
done
Part of what this script does is modify the input file (config.txt) to
account for residues that the user specifies under listVar. It does this
by accessing the 22nd line and replacing what is there with a new residue
number via starting_point_residue $RESNUM. After modifying the file and
resaving it, the program is run. The program output files are then saved to a
newly generated subfolder, named using the specified residue number.
22c portion of the sed command, based on its new
line number.CAVER Analyst
Once a structure is loaded in (File → Open Structure),
follow Tunnel → Computation. From there you can enter all
of the criteria for a tunnel search.
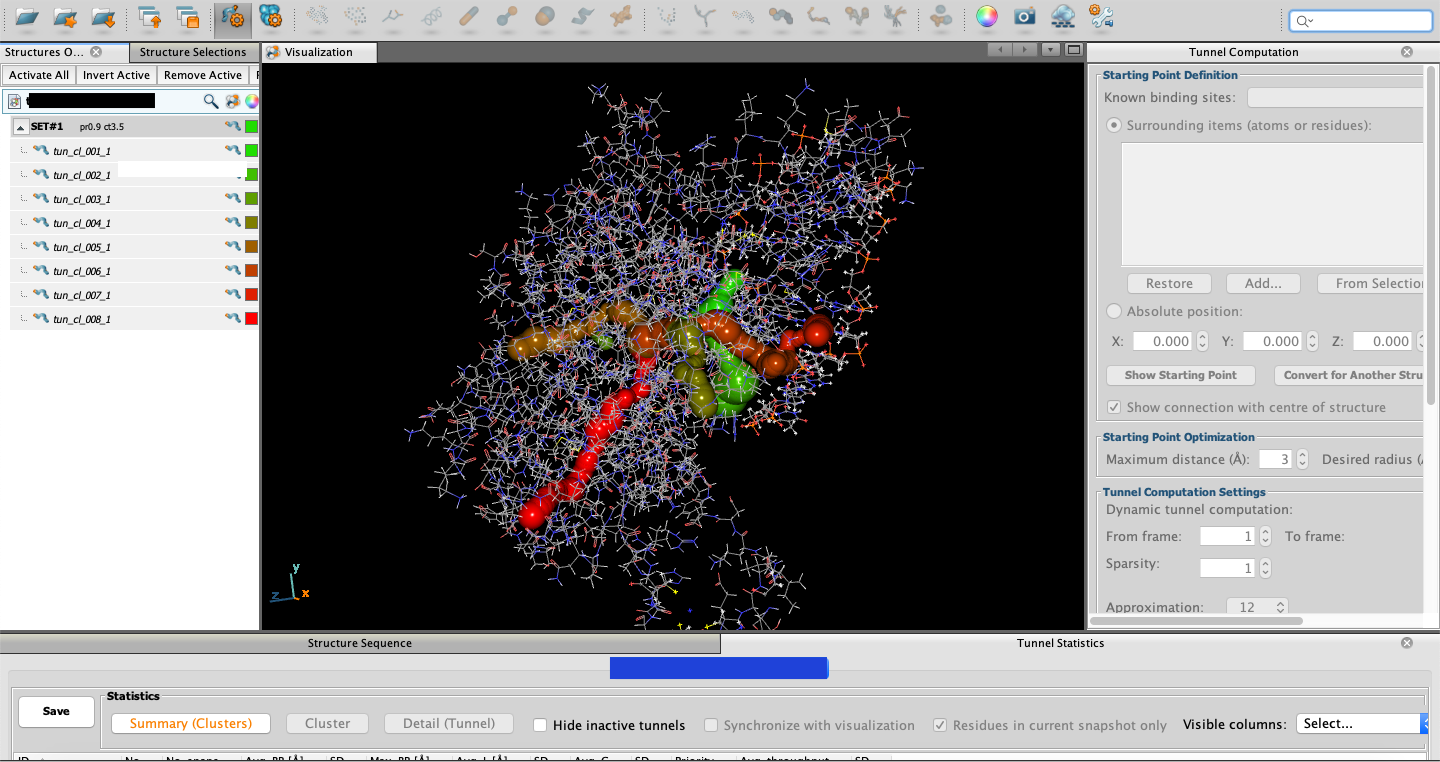
Under Known binding sites, a specific residue or atom can be given
using the PDB’s residue or atom number. It is suggested that you use a cofactor
or inhibitor for this criterion.
PyMOL plugin
PyMOL is a molecular visualization software. To use PyMOL for academic research, you need to purchase a license. More information on that is available on their website.
Once both PyMOL and the plugin are installed, you can use CAVER interactively. The information from the command-line input file becomes fill-in-the-blank boxes.