xleap is a GUI version of AMBER’s LEaP program. However, it can be deeply frustrating to work with for a few reasons. First, you cannot select a specific cursor position–you must backspace any previous parts of a command that you’ve made mistakes in typing. Second, you cannot have numlock enabled when using xleap because of various components in the edit module. Third, you can’t copy and paste commands into xleap. I’m sure by now you see why it can be a painstaking module to use. Why use it at all, then? xleap enables you to check what you’ve done, and make clear modifications. Sometimes your prepi files are failures, but have enough right that you don’t want to forcibly fix them. You can redraw bonds, move atoms, and more in the editor. In the words of Alice, “only LEaP knows what LEaP wants.” If it’s right in LEaP, then it’ll be right in your AMBER simulations, even if your solvated PDB looks incorrect in VMD (obviously recheck it after minimization–it should look correct in VMD by then).
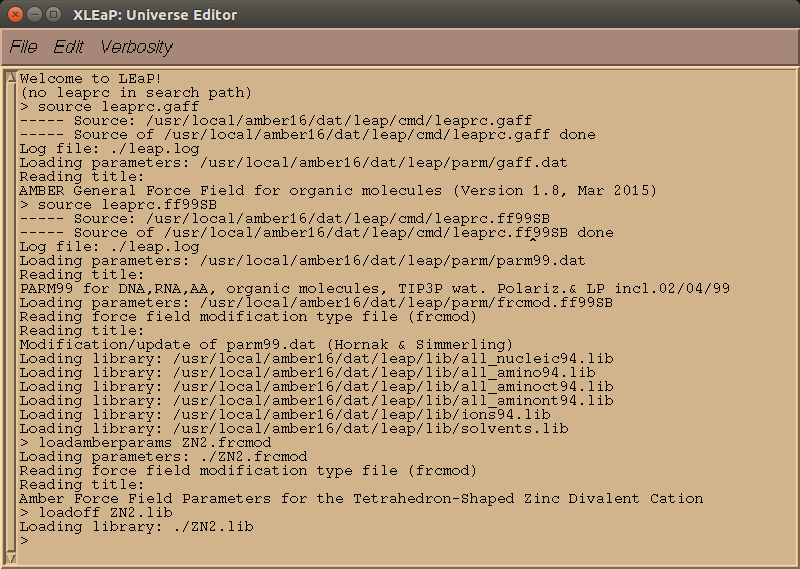
xleap uses the same command syntax that was seen with tleap (see the table). To open the program, open a Terminal, change directories to the folder with the files you need to set-up the system (so PDBs, prepi files, frcmod files, mol2 files, and lib files), and enter
$ AMBERHOME/bin/xleap
into the command line.
ssh -X or ssh -Y when you began the connection).
After doing this, the GUI will pop up. The Terminal is now useless until you
close xleap through (a) typing quit in its command line,
(b) clicking the orange X (not recommended, it’s not the safe way to quit), or
(c) following File → Quit.Once the GUI appears (see below), you can start entering the commands to set up your system.
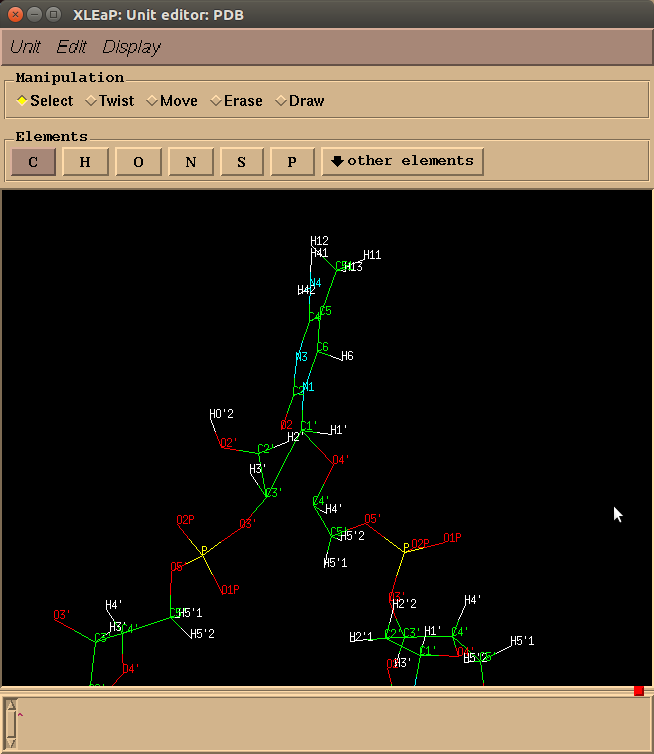
Individual components can be edited and checked in xleap after all the
necessary things have been added. For a loaded PDB, individual residues
(and their connections) can be edited using the name it was loaded in with
(ex. select PDB.123 would pick residue 123).
These different components can then be edited with something like edit PDB,
which will open the Unit editor.
Remember, the editor will not work properly if numlock is enabled, so make
sure that that is toggled off.
Following Edit → Show selection only will show the residues you
highlighted. The highlighting can be turned off by typing something
like deselect PDB in the command line Universe Editor.
To zoom inside the Unit editor, hit the Ctrl key while simultaneously
right-clicking and dragging.
Pressing the Ctrl key and moving the mouse will reorient the system.
You can translate the system with just right-clicking, like you would in VMD.
The atom names and types can be displayed by using the options under the
Display heading.
Highlighting an atom in the editor box will select it
(provided Select is marked under Manipulation) and choosing the other
effects under Manipulation can adjust it.
A system that needs editing in the Unit editor is shown below.
Every system is different, and each has its own little quirks. There are general trends, though, and things that should be saved along the way. An example of system set up with xleap is:
> source leaprc.gaff
> source leaprc.protein.ff14SB
> loadoff atomic_ions.lib
> addAtomTypes { { "DZ" "Zn" "sp3" } { "Zn" "Zn" "sp3" } }
> loadamberparams ZN2.frcmod
> loadoff ZN2.lib
> check ZN2
> loadoff akg.lib
> check AKG
> loadamberprep NonStandardA.prepi
> loadamberparams NonStandardA.frcmod
> check NSA
> NSB = loadmol2 NonStandardB.mol2
> loadamberparams NonStandardB.frcmod
> check NSB
> list
> PDB = loadpdb wildtype-system-AB.pdb
> check PDB
> savepdb PDB wildtype-system-AB-vac.pdb
> saveamberparm PDB wildtype-system-AB-vac.prmtop wildtype-system-AB-vac.inpcrd
> solvatebox PDB TIP3PBOX 12.00
> addions PDB K+ 0
> savepdb PDB wildtype-system-AB-wat.pdb
> saveamberparm PDB wildtype-system-AB-wat.prmtop wildtype-system-AB-wat.inpcrd
> quit
mol2 saying
that the parameters cannot be found, make sure that the argument you loaded
it in using (NSB above) matches the 3-letter code for the non-standard.