First things first: the AMBER LEaP tutorial is incredibly explanative. LEaP, in the forms tleap or xleap is used to generate AMBER systems.
Table: Commands in the LEaP syntax
| Command | Objective | Example | Ex. Explanation |
|---|---|---|---|
| source | load in force field parameters | source leaprc.ff14SB |
loads in the ff14SB force field |
| loadpdb | load in a PDB file | loadpdb foo system.pdb |
loads in the PDB, all future references are to foo |
| loadmol2 | load in a mol2 file | res = loadmol2 residue123.mol2 |
loads in the topology and charge information for non-standard residue, all future references are to res |
| loadamberprep | load in a prepi file for a non-standard residue | loadamberprep residue234.prepi |
loads in the topology information for the non-standard residue file residue234.prepi |
| loadamberparams | load in an frcmod file for a non-standard residue | loadamberparams residue234.frcmod |
load in the force field information for the non-standard residue file residue234.prepi |
| check | make sure that there aren’t errors | check foo |
checks the loaded foo for errors |
| select | choose specific atoms for the Unit editor | select foo.135 |
selects residue 135 of foo |
| edit | opens the selection in the Unit editor | edit foo |
opens foo the Unit editor; any selected residues will the be highlighted |
| solvateoct | solvate the system as a truncated octahedron | solvateoct foo TIP3P 12.00 |
solvates the loaded foo with TIP3P water extending at least 12.00 Å from the protein’s surface |
| solvatebox | solvate the system as a square box | solvatebox foo TIP3P 12.00 |
solvates the loaded foo with TIP3P water extending at least 12.00 Å from the protein’s surface |
| addions | add ions to neutralize the system (commonly K+, Na+, or Cl-) | addions foo K+ 0 |
neutralizes foo with potassium ions to a net charge of 0 |
| saveamberprep | saves a prepi file | saveamberprep R234 res234-fix.prepi |
saves a new prepi file, which means that fixed systems can be rebuilt with the modified prepi |
| saveamberparm | save the parameter and topology file | saveamberparm foo system_wat.prmtop system_wat.inpcrd |
saves the parameter and topology files for foo that will be used for simulation |
| savepdb | saves a PDB file | savepdb foo system.pdb |
saves the PDB file for foo system that can serve as an informative reference |
| quit | exit out of the program | quit |
you guessed it… it quits |
With LEaP, there are several solvent shapes to pick from.
We commonly use periodic solvent boxes, which are generated using either
solvateOct or solvateBox (both shown in the figure below).
solvateOct solvates the system in a truncated octahedron and solvateBox
solvates the system in a cuboid box.
The solvateOct command makes space-filling spherical shape.
This reduces solute rotation and often results in smaller systems.
Having a smaller system can save time in simulations.
Occasionally solvateOct has issues with centering itself correctly,
but those are few and far between.
Choose the solvation command you want and be consistent across that project.
Additional details on these commands can be found on page 232 of the
Amber18 Manual.
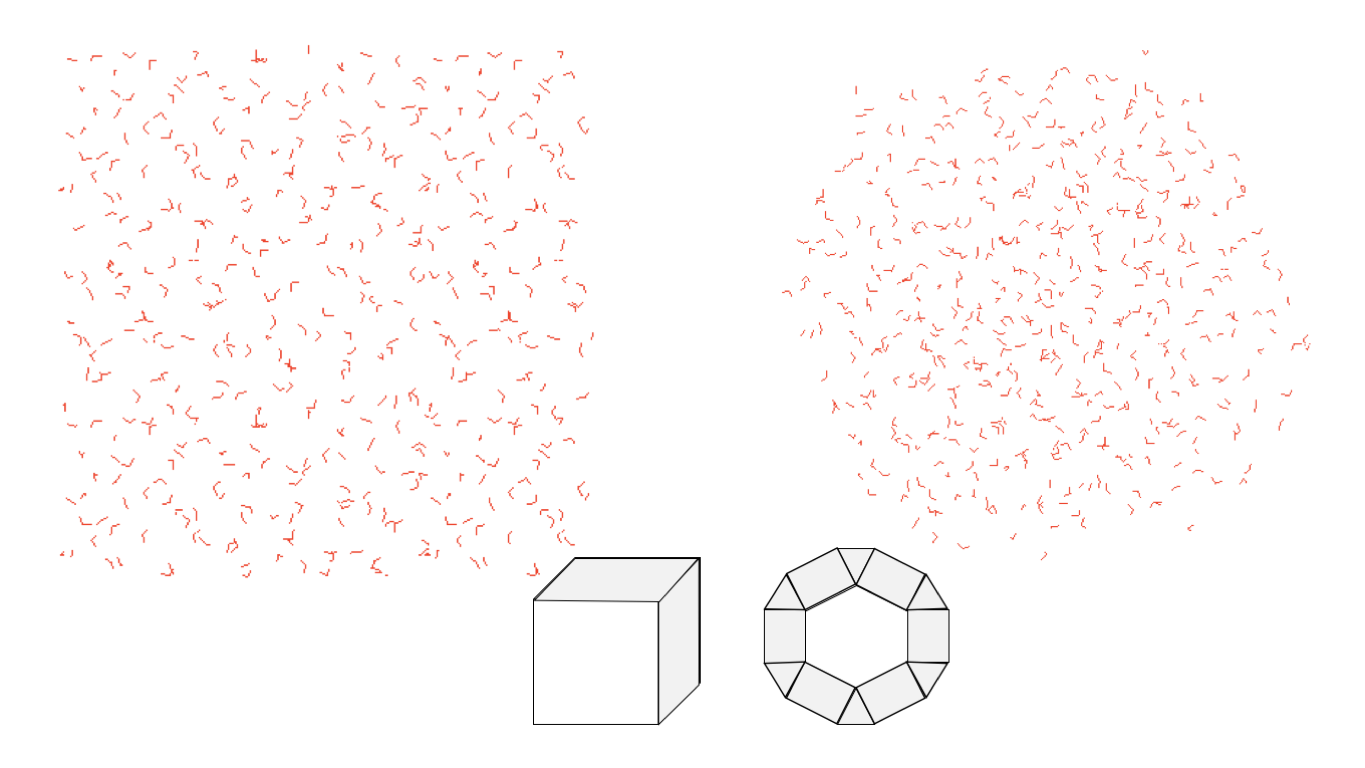
solvateBox (left) and solvateOct (right).