If you’ve never taken general chemistry, then the title here probably makes zero sense. If you have taken general chemistry, then the words “multiple protonation states” might spark panic. Never fear! With help of the Internet (and a little ~chemical intuition~), our systems will be biologically relevant!
So what do I mean by multiple protonation states? You’ve probably heard of (and used) ammonia at some point. Ammonia is NH3. Its conjugate acid, an ammonium ion, is NH4+. They differ by–you guess it–a single hydrogen. They’re pretty similar things, and whether or not that hydrogen is there is pH-dependent.
Why should you care? Certain amino acids, like histidine (sigh), can have multiple protonation states. That’s actually a good thing, because then you get stuff like acid-base catalysis. But that also means things like histidine can be the difference between accurate or garbage simulations.
There are five amino acids that can have weird protonation states. They are: aspartate, cysteine, glutamate, histidine, and lysine.
Figure: Protonation States
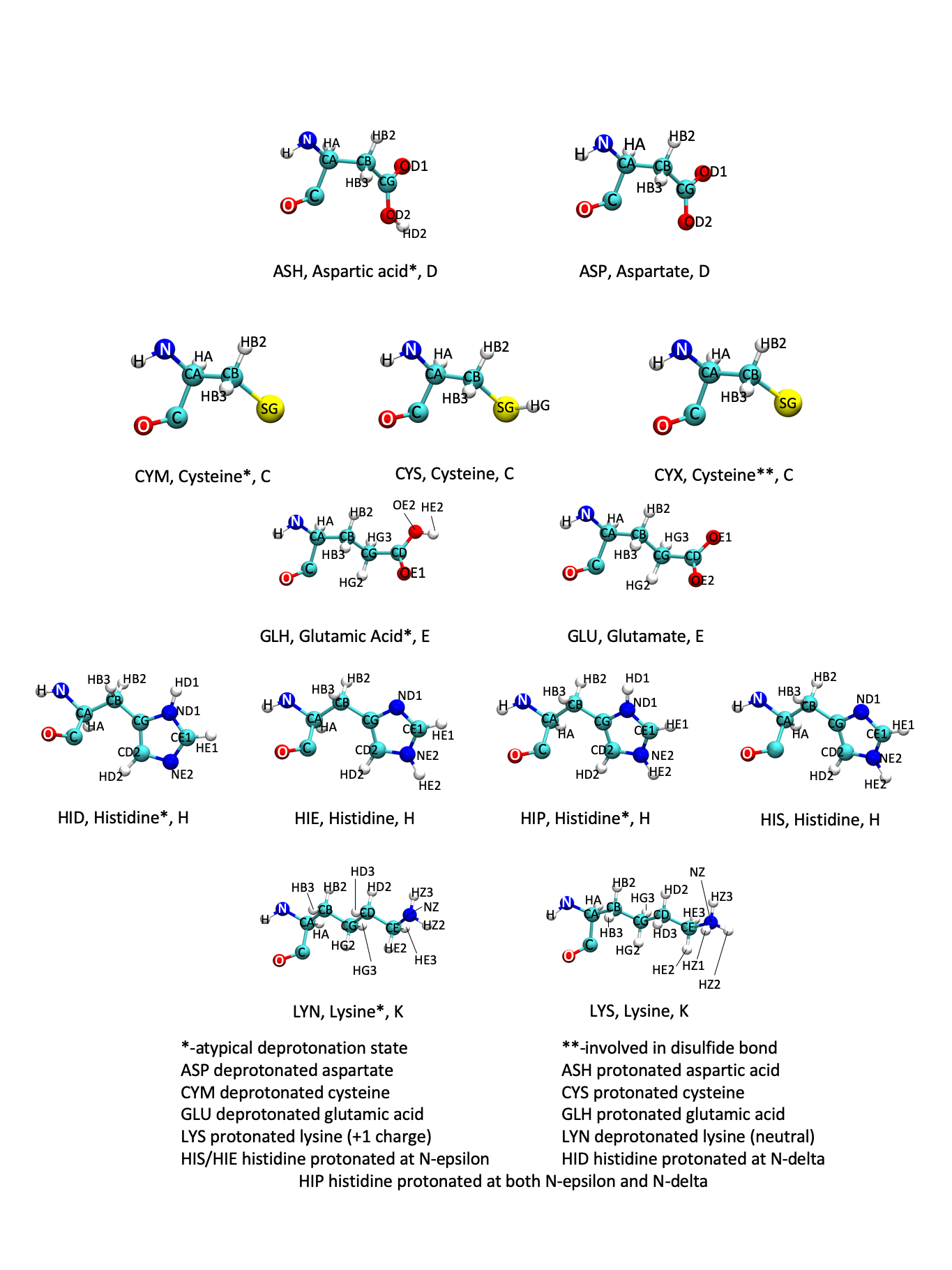
There are a few ways to ensure that your protein is protonated correctly. These are described below.
PROPKA (or the web version PDB2PQR)
is a program intended to predict the correct protonation states of protein
residues.
When a PDB file is downloaded from RCSB, there are no
hydrogens.
This is because hydrogens move too quickly and have so little mass that they
cannot be resolved using the determination method (e.g., x-ray crystallography).
Most MD programs will interpret residue names literally, which means that HIS
will be interpreted as a singly-protonated histidine residue.
However, that histidine residue could be protonated in other ways, such as
doubly-protonated or oppositely singly-protonated
(see the residue definitions here for more
information).
That’s why it is important to use something like
PROPKA or
the H++ webserver
to properly protonate the system.
Cleaning the Starting Structure
The PDB downloaded from RCSB has a lot of information in it (as mentioned on the PDB page). There’s a lot of stuff that is important for crystallographers that is honestly not well-taught to computational chemists. Luckily, RCSB created a website called PDB 101 that can explain a lot of that stuff.
As part of the PDB, there are often multiple chains.
Sometimes you need multiple chains (like in hemoglobin) for a complete system.
If there are multiple chains, there is still a chance you don’t need more than
one–often, symmetric dimers will be simulated as a monomer because it cuts
down on the computational cost.
There are assuredly times when you need the dimer, like if you want to study
non-symmetric mutations, but that’ll be fleshed out when you’re preparing
(or re-preparing) your structure.
These chain IDs come after the residue name (e.g., ACE A).
Before the residue name, there will occasionally be additional letters
(e.g., AACE).
These letters specify that there are alternate conformations for that residue.
The REMARK lines would provide more guidance as to how much either of those
conformations are favored.
Usually, using A is fine, but it’s better to spend more time investigating
at the beginning than to have to redo everything.
The following script (clean-pdb.sh) will read in the PDB downloaded from
RSCB.
Only the relevant lines will be read (i.e., REMARK likes will be ignored).
Any B-conformations will be removed (only A will be saved).
Water lines labeled HOH will be renamed WAT.
The final lines that are commented out would get rid of common inhibitors.
You can check the PDB’s RCSB page to see what small molecules were included.
The paper hopefully published with the structure would explicitly state which
inhibitor was used.
#!/bin/bash
## Define your files
thing=RCSB.pdb
thing_clean=RCSB_clean.pdb
## Clean the RCSB PDB (Remove crystal junk)
grep -e '^ATOM\|^HETATM\|^TER\|^END' $thing > $thing_clean
## The remainder will only act on lines containing the specified info
## Remove any B chain lines
sed -i '/BALA/d' $thing_clean
sed -i '/BARG/d' $thing_clean
sed -i '/BASN/d' $thing_clean
sed -i '/BASP/d' $thing_clean
sed -i '/BCYS/d' $thing_clean
sed -i '/BGLU/d' $thing_clean
sed -i '/BGLN/d' $thing_clean
sed -i '/BGLY/d' $thing_clean
sed -i '/BHIS/d' $thing_clean
sed -i '/BILE/d' $thing_clean
sed -i '/BLEU/d' $thing_clean
sed -i '/BLYS/d' $thing_clean
sed -i '/BMET/d' $thing_clean
sed -i '/BPHE/d' $thing_clean
sed -i '/BPRO/d' $thing_clean
sed -i '/BSER/d' $thing_clean
sed -i '/BTHR/d' $thing_clean
sed -i '/BTRP/d' $thing_clean
sed -i '/BTYR/d' $thing_clean
sed -i '/BVAL/d' $thing_clean
## Rename A chain as only chain
sed -i 's/AALA/ ALA/g' $thing_clean
sed -i 's/AARG/ ARG/g' $thing_clean
sed -i 's/AASN/ ASN/g' $thing_clean
sed -i 's/AASP/ ASP/g' $thing_clean
sed -i 's/ACYS/ CYS/g' $thing_clean
sed -i 's/AGLU/ GLU/g' $thing_clean
sed -i 's/AGLN/ GLN/g' $thing_clean
sed -i 's/AGLY/ GLY/g' $thing_clean
sed -i 's/AHIS/ HIS/g' $thing_clean
sed -i 's/AILE/ ILE/g' $thing_clean
sed -i 's/ALEU/ LEU/g' $thing_clean
sed -i 's/ALYS/ LYS/g' $thing_clean
sed -i 's/AMET/ MET/g' $thing_clean
sed -i 's/APHE/ PHE/g' $thing_clean
sed -i 's/APRO/ PRO/g' $thing_clean
sed -i 's/ASER/ SER/g' $thing_clean
sed -i 's/ATHR/ THR/g' $thing_clean
sed -i 's/ATRP/ TRP/g' $thing_clean
sed -i 's/ATYR/ TYR/g' $thing_clean
sed -i 's/AVAL/ VAL/g' $thing_clean
## Rename HOH as WAT
sed -i 's/HOH/WAT/g' $thing_clean
## Optional: Delete inhibitor/artifact lines
## Check the "Small Molecules" section of RCSB
## And compare against paper/common inhibitors for enzyme class
#sed -i '/ACT /d' $thing_clean
#sed -i '/OGA /d' $thing_clean
#sed -i '/SO4 /d' $thing_clean
#sed -i '/GOL /d' $thing_clean
#sed -i '/FSU /d' $thing_clean
#sed -i '/EDO /d' $thing_clean
PROPKA
The PDB2PQR web version of PROPKA
has a list of selections to make.
By default, it uses the PARSE forcefield, but you will likely want to select
AMBER.
Similarly, you will want to pick an output naming scheme, which will again
likely be AMBER.
The remaining things are preselected, and they should be fine:
- Ensure that new atoms are not rebuilt too close to existing atoms
- Optimize the hydrogen bonding network
- Create an APBS input file (this also enables the option to run APBS and
visualize your results through the web interface, if it has been installed)
Finally, there are pKa options. You should be able to use
pH 7, but if there was a crystal structure paper for the PDB and that has a pH listed, you may want to consider using that pH. Keep the default “Use PROPKA to assign protonation states at provided pH” selection.
Once you’ve submitted the job, download the files.
The PQR file will contain the residues with differing protonation states.
The following script (ph_changes_propka.sh) will select all these lines
and save them to a new file.
#!/bin/bash
## Define your files
thing=RCSB.pqr
thing_clean=RCSB_ph_list.pqr
## Get the non-standard protonation states
grep -e ‘ASH\|CYM\|CYX\|GLH\|HID\|HIP\|LYN’ $thing > $thing_clean
That didn’t have to be a script, but it seemed like it would be annoying to type out in the future.
You can then copy these non-standard protonation lines into the original PDB.
After using PROPKA, you can then use MolProbity to add missing hydrogen atoms and check whether ASN, GLU, and HIS residues need to be flipped.
H++
The H++ webserver can also be used to determine the protonation state of titratable residues. It has a few additional advantages, including checking if ASN, GLU, and HIS should be flipped and adding all missing hydrogen atoms. This means H++ is more of a “one stop shop” for PDB preparation.
To use it, select the “Process a Structure” tab.
Figure: H++ Upload
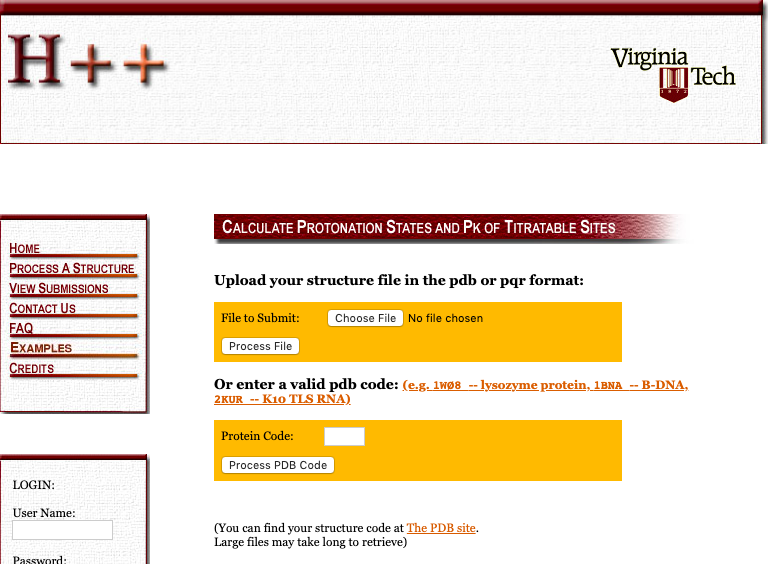
Process a Structure page for H++.Then, submit! After submission, you get to select some options.
Figure: H++ Options
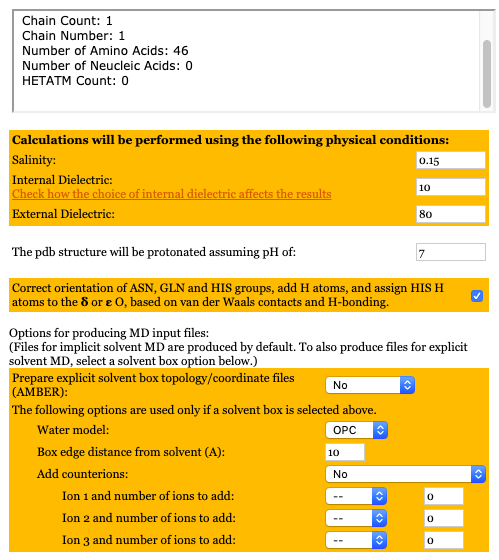
You will immediately get the following error message if there are missing loops on non-standard residues:
THE CALCULATION HAS STOPPED. It appears that some residues are missing in the middle of the uploaded
structure. When residues are missing (that is the sequence in the PDB file is discontiguous) the
accuracy of pK estimates of any kind is affected, especially in the vicinity of the gaps. Make sure
there are no "gaps" in your structure, see the FAQ for suggestions. It is also possible that your
structure contains non-standard residue names, sometimes listed as HETATM, in the main sequence.
These will not be recognized by the system and will be treated as missing, resulting in the above
error. Please change the names to comply with the standard amino-acid nomenclature. Alternatively,
you can supply your own PQR file -- the naming compliance is not enforced then. See the FAQ.
The added benefit of H++ is that it could be used to solvate and neutralize the system.