Python Basics
Overview
Teaching: 35 min
Exercises: 25 minQuestions
What are Python packages?
What are comments and docstrings?
How do I use variables?
What are if statements, for loops, and counters?
How do I write a function in Python?
Objectives
Create if statements and for loops.
Write a Python function.
Describe common formatting rules for Python.
Import Python packages.
Highlights
- Running Python Code
- Python Packages and the
importCommand - Additional Resources for Getting Started with Python
- Comments and Docstrings
- Data Types
- Trouble Spot: Integers and Floats
- Variables
- Print Statements and If Statements
- Python 3 Print Statements are Finicky
- For Loops and Counters
- Combining Ifs and Fors
- One Solution for Combining Ifs and Fors
- Functions
- Pizza Cost
- One Solution for Pizza Cost
- Importing and Using
numpy - Python starts indexing at 0 (zero).
Running Python Code
Python code can be run using a Python prompt (likely evoked through python3,
but that might be different depending on what versions are installed on your
device).
If you’re running a Jupyter Notebook or a Google CoLab Notebook, code
cells will run in the default Python shell (and can be changed to specific
versions).
user@host$ python3
Python 3.7.3 (default, Mar 27 2019, 16:54:48)
[Clang 4.0.1 (tags/RELEASE_401/final)] :: Anaconda, Inc. on darwin
Type "help", "copyright", "credits" or "license" for more information.
>>>
To run a Python script, you would use that specific prompt command with the name of the script you are trying to run.
user@host$ python3 my_cool_script.py
If you’re not sure what the Python you’re using, but you’ve installed Anaconda3,
go to /anaconda3/bin and see which executables are there.
If you’re having issues running the code provided, but know you have Python3
installed, run which python to see what version is being sourced.
It is not recommended that you change your default Python installation,
because this can have serious repercussions for your operating system.
Instead, save it to an alias that you call when you need to run Python3.
Python Packages and the import Command
Python has some basic commands (referred to lovingly as “base Python”), but
those are often not enough for what you want to to with Python.
Packages (or modules or libraries) are bundles of code that are designed for
a specific purpose.
They’re like a package in a mail sense–if you open a box you got in the mail,
the box was was way to wrap various objects into a neat little unit for shipping.
For instance, matplotlib is a library built for data visualization.
Using packages is similar to how module load works on an HPC cluster.
When you need to use a package for a specific purpose, you need to
import it into Python.
Additional Resources for Getting Started with Python
Comments and Docstrings
If you’re familiar with writing code, hopefully the importance of adding
comments has been stressed to you.
If this is your first time dabbling in coding, comments are arguably more
important than the code itself.
Comments provide information and reasoning for the actual code itself.
They are offset from the rest of the code with a specific symbol (or symbols).
In Python, a comment is made with the # (pound/hashtag/octothrope) symbol.
# This is my first comment
The nice thing about Python is that you can stack those symbols into your own
personal commentary style, or use the # symbol to create fancy comment blocks.
## I comment almost everything with 2 # symbols. It makes it clear to me that
## it's a comment.
## Note: the # symbol is only for single-line comments, so each line needs
## its own mark!!!!
#-----------------------------------------#
# Insert pretty comment block #
#-----------------------------------------#
Multiline commenting is possible in Python, but through something known as a docstring. They’re called docstrings because they are strings used for documentation purposes. Most projects (open and not) require coders to write docstrings for their functions, though they may have their own formatting for those docstrings. A docstring is generated through either three single or double quotes that both begin and end the docstring.
"""
This is a docstring
"""
'''
This is also docstring
'''
"""I can even do them in-line, but I don't know what I'd want to."""
'''The only rules are that you be consistent and escape characters other times
that you\'d want them.'''
The last segment of the above example talks about using an escape character.
In Python, the escape character is \.
Data Types
There are a number of different data types in Python, but a few robustly used types. This is by no means an exhaustive list, as new data types can be created.
| Data Type | Common Word | Brief explanation |
|---|---|---|
| str | string | Strings are things like filenames or text. x = "Thing" |
| int | integer | A typical integer. x = 1 |
| float | float | Floats are real numbers. x = 1. and x = 1.0 are both floats. |
| bool | boolean | True or False |
| dict | dictionary | Useful for holding data that needs to be paired, like school and abbreviation. These are known as key-value pairs. |
| list | list | Similar to an array, but of undefined length. They can be changed. |
| tuple | tuple | Like a list, but cannot be changed. |
The type() command will return the type of a variable.
x = 5
type(x)
<class 'int'>
There are times where you need to change a data type, like needing a number input option to be read as an integer. This is done with the “Data Type” specified above. You can either resave the value of that variable as the new type, or use it as part of a different command for a temporary change.
x = "5"
x = int(x)
y = 1
y = float(y)
Trouble Spot: Integers and Floats
One easy way to get tripped up in Python is to use an integer when a float is needed and vice versa. If you input
1., then you need a float input, and Python will complain about receiving an integer input.>>> x = 5. >>> for i in range(x): >>> print(i) Traceback (most recent call last): File "<stdin>", line 1, in <module> TypeError: 'float' object cannot be interpreted as an integerNow, it will render as expected by forcing
xto be an integer for the loop.>>> for i in range(int(x)): >>> print(i) 0 1 2 3 4
Variables
Variables are your friend! They’re a means to store data or information.
In Python, you typically just make a named object = before whatever you’re
trying to store as a variable.
x = "Thing"
print(x)
Thing
Print Statements and If Statements
Printing is useful for so many things in coding, from giving you the result to sharing information at specific checkpoints in the code.
In Python, the command to print is simply print.
print("Hello world")
Hello world
Python 3 Print Statements are Finicky
Python changed substantially between Python 2 and Python 3. In Python 2, you could make print statements without parenthesis. In Python 3, however, this will trigger an error message.
print x,yFile "<stdin>", line 1 print x,y ^ SyntaxError: Missing parentheses in call to 'print'. Did you mean print(x,y)?
We can use an if statement to print things only when a specific condition is
met.
These statements are often used with booleans, meaning variables marked as
True or False.
On days that you’ve had your morning coffee, you are in a decent mood.
But the days that start without coffee can leave you cranky.
This can be written as an if statement.
Python is very particular about indentation levels, and by convention, and
indent should be 4 spaces.
That said, as long as you’re consistent throughout, tabs can be used.
morning_coffee = True
if morning_coffee == True:
print("Hello world! What a wonderful day it is!")
else:
print("I'm not talking to you until I've had my morning coffee.")
Hello world! What a wonderful day it is!
If we reset the morning_coffee variable to False, we get a different output.
morning_coffee = False
if morning_coffee == True:
print("Hello world! What a wonderful day it is!")
else:
print("I'm not talking to you until I've had my morning coffee.")
I'm not talking to you until I've had my morning coffee.
We aren’t limited to booleans, which makes our if criteria more complex.
Grades can be assigned using an if statement.
final_grade_percent = 87.4
if final_grade_percent >90. and <=100.:
final_letter_grade = 'A'
elif final_grade_percent >80. and <=90.:
final_letter_grade = 'B'
elif final_grade_percent >70. and <=80.:
final_letter_grade = 'C'
elif final_grade_percent >60. and <=70.:
final_letter_grade = 'D'
else:
final_letter_grade = 'F'
print("The final percent grade of {} yields {} as a letter\
grade.".format{final_grade_percent,final_letter_grade})
The final percent grade of 87.4 yields B as a letter grade.
Some notes about what happened there:
- Integers differ from floats with the extra
. - The
\character is a line continuation mark. It is a good practice to keep lines under 80 characters. - Python will complain if the
:(colon) is not there.
elif and else are not required for every if statement.
Say we had a Python script that wrote to the file file123.txt.
Users that use a file named file123.txt as an input, will overwrite their
input file with the script’s output, and that could be frustrating.
So, we may want to give the output file a different name, but only when the
input name matches what the output name would otherwise be.
# Pretend that output_file = "file123.txt" is up here
# somewhere where we don't see it and aren't planning on editing it
input_file = "some_input.txt"
if input_file == "file123.txt":
output_file = "output_file123.txt"
print(output_file)
output_file = file123.txt
Another important word is continue.
continue is great if you know when you don’t want something to happen in
one specific case, but you do need it for every other case.
## Give a mouse a cookie, but only if they haven't already have a cookie
if cookie == True:
continue
else:
cookie == True
Similarly, if you need to exit out of the statements and move on in the code
when something specific happens (like receiving an input that would crash a
program), you can use break.
For Loops and Counters
for loops control iterations.
In Python, for loops start use indents to keep the information within the
loop contained.
Typically, these look like for x in y:.
Again, Python will complain if there isn’t a colon.
coin_list = ["quarter", "dime", "nickel", "penny", "half-dollar",
"golden dollar"]
for coin in coin_list:
print(coin)
quarter
dime
nickel
penny
half-dollar
golden dollar
In the above example, the for loop was initiated with for coin in coin_list:.
However, the use of coin is irrelevant–it was just a placeholder variable.
Many people like to use i for their for loop indexing, and it could have
just as easily been for i in coin_list.
However, in that case, swapping out coin for i must also be done within
the for loop itself.
Otherwise, you’ll get a name is not defined error.
>>> coin_list = ["quarter", "dime", "nickel", "penny", "half-dollar",
... "golden dollar"]
>>>
>>> for i in coin_list:
... print(coin)
...
Traceback (most recent call last):
File "<stdin>", line 2, in <module>
NameError: name 'coin' is not defined
The other common initiation of a for loop in Python is with the range()
command.
for x in range(5):
print(x)
0
1
2
3
4
Sometimes it is necessary to keep a count within code, or otherwise update
a variable through addition or substration.
After setting a variable to an initial value, you can update it with +=
or -=.
This means that x += 1 can take the place of x = x + 1 (spaces optional).
y = 0
for x in range(5):
y += 1
print(y)
5
Combining Ifs and Fors
Write a for loop that reads in a list of 5 items and does not print anything for one of them.
One Solution for Combining Ifs and Fors
There are many, many ways to write code. This is just one solution.
MD_programs = ['AMBER', 'CHARMM', 'GROMACS', 'LAMMPS', 'TINKER'] for program in MD_programs: if program == "TINKER": continue else: print("You don't have to convert from {} format to XYZ. Yet.".format(program))You don't have to convert from AMBER format to XYZ. Yet. You don't have to convert from CHARMM format to XYZ. Yet. You don't have to convert from GROMACS format to XYZ. Yet. You don't have to convert from LAMMPS format to XYZ. Yet.
Functions
def stands for definition. Callable functions are created with the def
command.
Whatever you name your variable in the function call does not have to be what
was in the definition line.
We can turn the earlier grades if statement into a function.
def assign_grade(final_grade_percent):
if final_grade_percent >90. and <=100.:
final_letter_grade = 'A'
elif final_grade_percent >80. and <=90.:
final_letter_grade = 'B'
elif final_grade_percent >70. and <=80.:
final_letter_grade = 'C'
elif final_grade_percent >60. and <=70.:
final_letter_grade = 'D'
else:
final_letter_grade = 'F'
return final_letter_grade
To later use this function, we would assign a value to final_grade_percent
and call
final_letter_grade = assign_grade(final_grade_percent)
Pizza Cost
Write a function that determines the cost of a pizza based off of the cost of individual toppings.
One Solution for Pizza Cost
There are many, many ways to write code. This is just one solution.
plain_pizza = 10. toppings = ['pineapple', 'pepperoni', 'anchovies', 'olives'] def pizza_price(plain_pizza, toppings): for topping in toppings: if topping == 'pepperoni': plain_pizza += 2. else: plain_pizza += 1. return plain_pizza plain_pizza = pizza_price(plain_pizza, toppings) print("The pizza costs ${:.2f}".format(plain_pizza))
Importing and Using numpy
One of the most common packags is numpy.
Python is really nice in that you can give these modules a “nickname” so it’s
easier to refer to them. You’ll often see np as a “nickname” for numpy.
Once it is loaded in, the functions that it contains are accessible using the
nickname (or the module name, if simply import numpy is used).
import numpy as np
x = 35
y = 493545435
thing = np.divide(x, y)
print("thing:", thing)
thing: 7.091545685150548e-08
The above code uses the numpy version of division, which may be faster or
give different significance depending on the operation being performed.
Division is something included in “base” Python (Python without any
imported modules). Because we’ve already defined x and y in a previous cell,
we can continue.
new_thing = x/ y
print(new_thing)
7.091545685150548e-08
In this case, the numbers are the same.
Python starts indexing at 0 (zero).
When working with arrays or lists, the very first position is not 1, but 0. This is arguably the most important thing to know about Python.
import numpy as np x_list = np.arange(0,10,1) print(x_list) for x in x_list: print(f"I am number {x} of {len(x_list)}.")[ 0 1 2 3 4 5 6 7 8 9] I am number 0 of 10. I am number 1 of 10. I am number 2 of 10. I am number 3 of 10. I am number 4 of 10. I am number 5 of 10. I am number 6 of 10. I am number 7 of 10. I am number 8 of 10. I am number 9 of 10.
Key Points
Python can be run interactively, through a script, or in a notebook form.
Use
importto load in Python packages.Comments are evoked through
#, and docstrings are evoked through""" """.Python starts indexing at 0 (zero).
Indentation levels are very important for Python, and 4 spaces should be used for indents. Indent levels are critical for
ifstatements andforloops.
ifstatements andforloops should each end the opening line with a:(colon).It is a common convention to use non-plural/plural pairs for
forloops.Updating a variable with addition can be achieved through
+=.The
\is a line continuation mark, as well as the escape character.The phrase keyword arguments (for functions) is sometimes shorted to
**kwargs.Exit command-line Python with
quit()orexit(), including the parentheses.
Structure Preparation
Overview
Teaching: 120 min
Exercises: 0 minQuestions
How do I make a structure biologically relevant?
Do I need to add hydrogens?
Where do parameters come from?
How do I add ions and solvate a system?
Objectives
Modify a crystal structure to reflect realistic protonation states.
Check a structure for clashes.
Create parameters for a small, organic molecule.
Generate a solvated and charge-neutral structure with LEaP.
Highlights
- Where are the files?
- PDB Structure Background
- Initial Processing
- Possible Error
- What in the world are all those
sedcommands for? - Hold up, why did we remove
GOLagain? - PROPKA
- MolProbity
- LEaP
- “Decently described” sounds sketchy…
- Using
tleap - Testimonial from a Grad Student: Read the Error Messages
Where are the files?
Check out the web
PDB Structure Background
The ATOM and HETATM records in a PDB file contain the relevant information
for getting started with computational simulations.
ATOM 1 N HIS 4 30.098 2.954 7.707 1.00 0.00 N
The records start with ATOM or HETATM.
Next is the atom index or atom ID number.
Each atom index is unique to that atom in a simulation, but before the structure
is fully prepared, these numbers will often be non-unique.
Next is the atom name. Atom names should be unique within a given residue.
So, for a methyl group with 3 hydrogens, those hydrogens would likely be labeled
as H1, H2, and H3.
After the atom name comes the residue name.
AMBER uses unique residue names for residues in different protonation states,
but not every program or force field follows this method.
Next is the residue number.
These should also be unique once the structure is fully prepared.
The following three numbers are the X, Y, and Z coordinates for the atom.
The next two numbers are the occupancy and the temperature factor, respectively.
RCSB has a good explanation of these in their
PDB-101
site, but they are not important right now.
Finally, the element symbol for the atom is listed.
Initial Processing
Download 5y2s.pdb from RCSB.
RCSB has a dropdown for “Display Files” and “Download Files.”
Choose the PDB format.
PDB files have a lot of crystallographic information that can confuse MD
programs. The important lines start with ATOM, HETATM, TER, and END.
The clean_pdb.sh file is a script that includes the following command to
extract these lines from the PDB file and save them in a new file.
grep -e '^ATOM\|^HETATM\|^TER\|^END' $thing > $thing_clean
To run the clean_pdb.sh script, use:
bash clean_pdb.sh
or
./clean_pdb.sh
Possible Error
If you try the second option (./clean_pdb.sh), you may see:
-bash: ./clean_pdb.sh: Permission deniedWhen scripts are initially written, they do not have the appropriate permissions to be run. This is actually an anti-malware strategy! You don’t just want commands to be able to run all willy nilly. That’s why you then need to use another command,
chmodto modify the file permissions and allow it to be executed (run).chmod u+x clean_pdb.sh
Running the clean_pdb.sh script creates the 5Y2S_clean.pdb file.
What in the world are all those
sedcommands for?Each of those
sedcommands is for in-place editing of the file. The first block removes “B chain” residues, the second block renames the “A chain” residues as the normal residue name, and the third block renamesHOHtoWATand removes theGOL, which is an inhibitor. PDBs often have “A chain” and “B chain” residues, which offer alternative occupancies for an atom. These occupancies are kind of like saying 65% of the day someone sits at a desk, but the other 35% of the day they lay in their bed. You would likely care most about the time spent at the desk, but there are also times that knowing they’re in bed is important. For the purpose of this tutorial, we’re taking all the A-chain residues and ignoring all the B-chain residues. Every system is different, and every structure is different, so it’s better to make a case-by-case decision on which of the chains to use for a residue.Hold up, why did we remove
GOLagain?There are a few common inhibitors for enzymes, and
GOL(glycerol) is often one of them. You can check for potential inhibitors by reading the paper published with the crystal structure (if one exists…). This is one case where reading the methods section is critically important.
PROPKA
The crystal structure doesn’t come with hydrogens. This is because they both rotate frequently, and they are very, very small compared to everything else, so they’re not actually captured. Because of that, hydrogens are placed based on an educated guess, but protonation states are pH dependent. Thus, we need to use something to predict the proper protonation states for each titratable residue. Here, we’ll use PROPKA.
Go to the PROPKA webserver and select PDB2PQR.
Upload the 5Y2S_clean.pdb file.
These options should be used with the webserver:
- pH 7.0
- Use PROPKA to assign protonation states at provided pH
- Forcefield: AMBER
- Choose output: AMBER
- Deselect Remove the waters from the output file
Once the job has completed, there are a number of files to download.
The relevant output files have the pqr, stdout.txt and stderr.txt
extensions.
The pqr file contains the adjusted residue names.
Not to skip ahead in the tutorial (that’s why these files are provided 😃),
but you can use MDAnalysis to convert the output PQR back to a PDB.
This is what the propka-reintegration.py script looks like.
import MDAnalysis as mda
propka_output="../2-propka_output/p09m0bn8cm.pqr"
pdb_out="5Y2S_ph7_protonated.pdb"
system = mda.Universe(propka_output, format="PQR", dt=1.0, in_memory=True)
## Remove the segment IDs (w/o this, `SYST` gets annoyingly printed by element)
for atom in system.atoms:
atom.segment.segid = ''
system.atoms.write(pdb_out)
A physical inspection of the .pqr file from PROPKA reveals these REMARK lines:
REMARK 5 WARNING: PDB2PQR was unable to assign charges
REMARK 5 to the following atoms (omitted below):
REMARK 5 2157 ZN in ZN 301
REMARK 5 2158 C in CO2 302
REMARK 5 2159 O1 in CO2 302
REMARK 5 2160 O2 in CO2 302
REMARK 5 2161 C in CO2 303
REMARK 5 2162 O1 in CO2 303
REMARK 5 2163 O2 in CO2 303
This means that PROPKA couldn’t process the ZN or CO2 residues.
Because of that, they weren’t included in the PQR, and thus were not in the
converted PDB.
Make a copy of the converted PDB to add the ZN and CO2 residues into.
cp 5Y2S_ph7_protonated.pdb 5Y2S_ph7_nsa.pdb
Now, you can copy the ZN and CO2 atom lines from 5Y2S_clean.pdb into
5Y2S_ph7_nsa.pdb.
As part of this:
- Change the temperature factor (the number after XYZ coordinates) to
0.00 - Remove the A segment ID
- Change
HETATMtoATOM
The 5Y2S_clean.pdb ZN line
HETATM 2157 ZN ZN A 301 14.442 0.173 15.262 1.00 3.61 ZN
becomes
ATOM 2157 ZN ZN 301 14.442 0.173 15.262 1.00 0.00 ZN
You can leave the atom ID (2157) as-is.
Further programs will renumber everything.
MolProbity
Upload the cleaned PDB file (5Y2S_ph7_nsa.pdb) with the corrected pH AMBER
types to MolProbity.
As part of the structure processing by MolProbity, all of the hydrogens are
removed.
These hydrogens must then be re-added with the “Add hydrogens” button.
Selecting this button brings up a new page with options.
Choose Asn/Gln/His flips as the method.
In this particular case, the 5Y2S structure comes from x-ray crystallography.
Thus, select Electron-cloud x-H for the bond length.
Then you can hit Start adding H >.
MolProbity found evidence to flip several residues.
Select the ones you’d like to flip with the check-boxes (all recommended)
before hitting Regenerate H, applying only selected flips >.
Because of the addition of hydrogens and flips, a pop-up appears.
Select yes to download the new structure now.
LEaP
LEaP works best with a PDB that’s as close to true form as possible. The new structure downloaded from MolProbity has some issues with it. We need to rectify these things:
TERlines should be between the protein and any metals, ions, or waters- Remove the
flipandnewmarks that MolProbity prints - Remove
USERandREMARKfrom PROPKA and MolProbity - Use atom names that are consistent with what LEAP expects
- In this case, change
OWforWATtoO
- In this case, change
This PDB file was cleaned up through by deleting the USER and REMARK heading
lines, as well as performing these string replacements with vi.
:%s/ flip//g
:%s/ new//g
:%s/ OW WAT/ O WAT/g
The other important thing we need to consider about this structure is its active site. The CO2 residue is not native to AMBER, so we need to get parameters for it. If this were a residue that had hydrogens (like a drug), then the anticipated hydrogens would need to be added before it was parametrized. Additionally, the ZN residue in the active site is one that would benefit from additional parameters. In this case, the ZN if 4-coordinate with 2 HID, 1 HIE, and 1 WAT. This is adequately described by ZAFF (which has its own tutorial).
Generating CO2 Parameters
Since CO2 is a small, organic molecule, it should be decently described by GAFF.
“Decently described” sounds sketchy…
Good! You picked up on that! It can be sketch. GAFF stands for General AMBER Force Field. That means it’s generalized for organic compounds that contain C, N, O, H, S, P, F, Cl, Br and I. So, if you have something that’s tricky, or that you’d want more tailored parameters for, you should use RESP fitting to compute them. One way to achieve this is to use pyRED.
To get the GAFF parameters, run antechamber.
Each of the flags gives the program information it needs to run.
| Flag | Purpose |
|---|---|
| -i | input file |
| -fi | file type of input file |
| -o | output file |
| -fo | file type of output file |
| -c | calculation type |
| -s | verbosity |
| -nc | net charge |
| -m | multiplicity |
antechamber -i CO2.pdb -fi pdb -o CO2.mol2 -fo mol2 -c bcc -s 2 -nc 0 -m 1
Ordinarily, after running antechamber, you run parmchk to make sure that
all of the necessary force field descriptors are present.
parmchk -i CO2.mol2 -f mol2 -o CO2.frcmod
This file contains no information, as everything required is covered by GAFF.
Incorporate ZAFF
ZAFF is the Zinc AMBER Force Field (read more). There are several parameter sets for for different 4-coordinate zinc complexes.
Looking at the 3D view on the RCSB website, you can identify that HID 94, HID 96, HIE 119, and WAT 455 are all coordinated to ZN 301 in the original structure. The 3 histidine, 1 water coordination is best described by ZAFF Center ID 4.
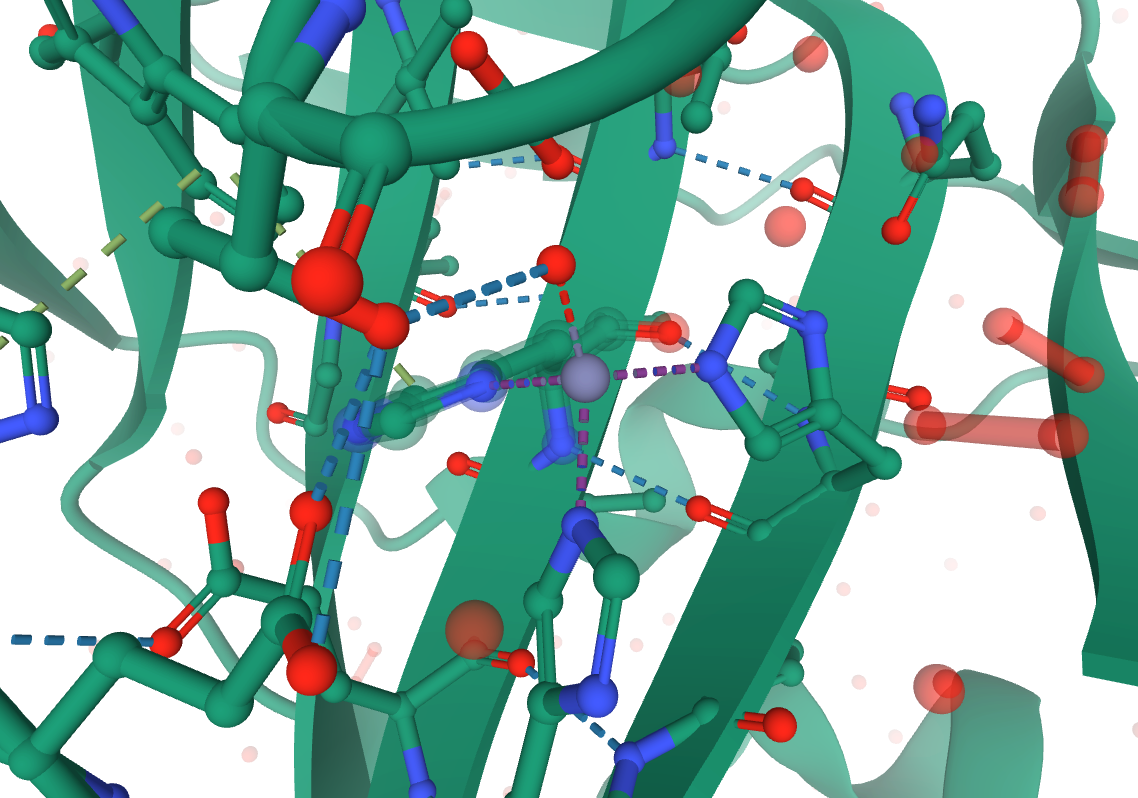
Image citation: 5Y2S, doi: 10.2312/molva.20181103, RCSB PDB.
Since PROPKA and MolProbity maintained the residue numbering, we can use those RESIDs to rename the coordinated residues as needed for ZAFF. Only the residue name (for the entire residue) needs to change.
| ResNum | Original ResName | New ResName |
|---|---|---|
| 94 | HID | HD4 |
| 96 | HID | HD5 |
| 119 | HIE | HE2 |
| 455 | WAT | WT1 |
| 301 | ZN | ZN6 |
Before moving on with LEaP, we need to download the parameter files for ZAFF. These can be downloaded from the ZAFF tutorial as a prep and frcmod. They are also included in the files downloaded for this lesson.
Using tleap
tleap is is the terminal form of LEaP.
There is also xleap, which uses a GUI.
Most of the time, people use an input script with tleap, which is evoked like:
tleap -f tleap.in
The tleap.in file for used here looks like:
source leaprc.gaff
source leaprc.protein.ff14SB
addAtomTypes { { "ZN" "Zn" "sp3" } { "N5" "N" "sp3" } { "N6" "N" "sp3" } { "N7" "N" "sp3" } }
source leaprc.water.tip3p
loadamberprep ZAFF.prep
loadamberparams ZAFF.frcmod
CO2 = loadmol2 CO2.mol2
c = loadpdb 5Y2S_ph7_mp_ZAFF.pdb
bond c.261.ZN c.94.NE2 ## HID NE2
bond c.261.ZN c.96.NE2 ## HID NE2
bond c.261.ZN c.119.ND2 ## HIE ND2
bond c.261.ZN c.318.O ## WAT O
savepdb c 5Y2S_vac.pdb
saveamberparm c 5Y2S_vac.prmtop 5Y2S_vac.inpcrd
addions c CL 0.0
solvatebox c TIP3PBOX 12.0
savepdb c 5Y2S_wat.pdb
saveamberparm c 5Y2S_wat.prmtop 5Y2S_wat_init0.rst
quit
First, the relevant force field information is sourced, and any new atom types
are defined.
Next, the information for ZAFF and CO2 are loaded in.
The CO2 = part corresponds to the 3-letter code that is in the PDB for that
residue.
Next, the protein is loaded in using a variable name, c.
Any variable name is fine, but if you update it, it’d need to be updated in
all of the remaining lines.
The bond lines generates a bond between the zinc and each of the atoms it
is coordinated to, which is an aspect of the ZAFF model.
The save lines save structures in various states.
There’s a first save before the ions and water are added, known as a “dry” or
“vacuum” structure.
Then, those ions (where 0.0 says to neutralize the charge) and waters are
added (where 12.0 specifies that water should extend 12.0 angstroms from the
protein’s surface).
These additions are saved in a “wet” or “solvated” structure.
LEaP does accept comments in these files, using #.
If you run this script on the provided files, it should work. However, if you were doing this on your own, you would likely encounter this error:
WARNING: The unperturbed charge of the unit: 0.998000 is not zero.
FATAL: Atom .R<NHIE 4>.A<HD1 20> does not have a type.
Failed to generate parameters
Parameter file was not saved.
Quit
The way to solve this error is to delete the HD1 hydrogen of the very first
residue, HIE 4, so that it can be rebuilt.
This is an error that can sometimes occur with first and last protein residues.
Testimonial from a Grad Student: Read the Error Messages
97.9% of my problems would be solved if I just read the error messages. Often, they tell you exactly what you need to do to fix it. If they don’t, I strongly recommend Google. Google is your best friend. Well, maybe not your best friend. I don’t even know you. I guess what I’m saying is that you’re probably not the first person to have an error of a specific type, which is why someone programmed an error message. Between the message, the internet, and the source code, there’s a lot of “clues” out there as to what’s going wrong.
Key Points
PROPKA is one way to calculate protonation states.
MolProbity can be used to check a structure for clashes, and make relevant ring flips.
Non-standard residues must have relevant hydrogens before parametrization.
For the final structure, LEAP will add hydrogens (and other missing sidechain atoms) based on residue definitions.
OpenMM
Overview
Teaching: 30 min
Exercises: 0 minQuestions
How do I set up an OpenMM molecular dynamics simulation in Python?
What information can OpenMM provide as the simulation is running?
Objectives
Load simulation inputs into Python.
Set up simulation parameters and environments.
Apply distance and angle restraints.
Set up outputs and reporters.
Run short trajectory of molecular dynamics.
Restarting a simulation from a checkpoint.
Highlights
- Importing OpenMM into Python
- Loading Simulation Inputs
- Setting up the Simulation Environment
- Running the Molecular Dynamics Simulation
- Restarting a Simulation from a Checkpoint
There is an OpenMM tutorial site with some additional “cookbook” style tutorials. This is meant as an introduction to the basics.
Importing OpenMM into Python
This portion assumes you have already installed the appropriate version of
OpenMM to your specific environment.
OpenMM is CUDA-compatible, and
different versions are available depending
on which version of CUDA you’re using.
import simtk as omm
from simtk.openmm.app import *
from simtk.openmm import *
from simtk.unit import *
from simtk.openmm import app
Loading Simulation Inputs
We assume you have already generated your .prmtop and .inpcrd files for your
system.
If you haven’t, use the example files provided.
Also, remember that assigning variables, classes, and objects in Python is
very important.
prmtop = app.AmberPrmtopFile("protein.prmtop")
inpcrd = app.AmberInpcrdFile("protein.inpcrd")
Setting up the Simulation Environment
OpenMM requires several pieces of information to get started, but assigning variables in stages can help keep things organized (and also easy to modify for future use.)
# Using Particle-mesh Ewald
system = prmtop.createSystem(nonbondedMethod = app.PME,
# with a 10 Angstrom cutoff distance
nonbondedCutoff = 1.0*nanometers,
# keeping bonds between hydrogens and heavy atoms at a fixed length
constraints = app.HBonds,
# setting the error tolerance for the Ewald
ewaldErrorTolerance = 0.001,
# letting waters be flexible instead of solid triangles
rigidWater = False,
# maintains the center of the periodic box at the center of mass
removeCMMotion = True)
You’ll also want to set up your thermostat to run in NVT (and add a barostat to run in NPT).
# Temperature, Friction Coefficient, Timestep
# This will be added to the larger simulation
thermostat = LangevinIntegrator(300*kelvin, 1/picoseconds, 0.001*picoseconds)
# Pressure, Temperature (only used for calculation),
# Frequency (how frequently the system should update the box size)
barostat = MonteCarloBarostat(1.0*atmosphere, 300.0*kelvin, 25)
# The barostat is added directly to the system rather than the larger
# simulation.
system.addForce(barostat)
If you need distance or angle restraints between atoms, you can add them here.
# Add distance restraints
distance_restraints = HarmonicBondForce()
# First atom index (Python starts at zero!), Second atom index, Distance,
# Force Constant
# You can use as many addBond() function calls as you like, if there are
# multiple distances to restrain.
# But be careful not to duplicate any, as they won't overwrite each other,
# just additively stack up.
distance_restraints.addBond(index1, index2, distance*angstroms,
force*kilocalories_per_mole/angstroms**2)
# Add the collection of distance restraints to your system.
system.addForce(distance_restraints)
# Add angle restraints
angle_restraints = HarmonicAngleForce()
# First, second, and third atom indices, with the second atom as the vertex of
# the angle, the desired angle in radians, and the force constant.
# As above, add as many angle restraints as you need, but be careful not to
# duplicate them.
angle_restraints.addAngle(index1, index2, index3, angle,
force*kilocalories_per_mole/radians**2)
# Add the collection of angle restraints to the system.
system.addForce(angle_restraints)
Now we need to set up the entire simulation with what we’ve done so far.
# The topology of the system
sim = Simulation(prmtop.topology,
# The system itself, with all the restraints, barostat, etc.
system,
# The thermostat, in this case the LangevinIntegrator we established before
thermostat)
# Gets the starting position for every atom in the system and
# initializes the simulation
sim.context.setPositions(inpcrd.getPositions(asNumpy=True))
# Establishes the periodic boundary conditions of the system
# based on the box size.
sim.context.getState(getPositions=True, enforcePeriodicBox=True).getPositions()
# Filename to save the state data into.
sim.reporters.append(StateDataReporter("output.log",
1000, # number of steps between each save.
step = True, # writes the step number to each line
time = True, # writes the time (in ps)
potentialEnergy = True, # writes potential energy of the system (KJ/mole)
kineticEnergy = True, # writes the kinetic energy of the system (KJ/mole)
totalEnergy = True, # writes the total energy of the system (KJ/mole)
temperature = True, # writes the temperature (in K)
volume = True, # writes the volume (in nm^3)
density = True)) # writes the density (in g/mL)
# Updates the checkpoint file every 10,000 steps
sim.reporters.append(CheckpointReporter(chk_out,10000))
# Adds another frame to the CHARMM-formatted DCD (which can be easily
# converted to .mdcrd by cpptraj) every 10,000 timesteps.
# It's good practice to keep the trajectory and checkpoint on the same
# write frequency, in case you need to stop a job and resume it later.
sim.reporters.append(DCDReporter("trajectory.dcd",10000))
Running the Molecular Dynamics Simulation
We have now set up the entire system for running molecular dynamics!
But first, as is good practice, we want to minimize the system first to clear
any potential clashes between atoms and residues.
# Minimize the system
sim.minimizeEnergy(maxIterations=2000)
You can set the maxIterations to whatever value you want, but be careful not to minimize for too long, as you can wind up starting from a structure that may be very different from your original files. From here, you can begin the actual MD portion.
# This instructs the program to take 1,000,000 timesteps, which corresponds
# to 1.0 ns of MD based on a 1fs timestep.
sim.step(1000000)
Be careful here, as this will write the entire trajectory to a single file as
it goes.
It’s a good idea to save backups along the way, especially if you’re
restarting from checkpoints.
Otherwise, the program will overwrite the file when you resume simulation,
and you can lose the data.
Restarting a Simulation from a Checkpoint
Sometimes we may want to pick up a simulation where we left off.
This requires only some slight modifications to the process.
Most of the script you built above can be used, with a few exceptions.
First, remove the following portion, as we no longer want the starting
positions determined by the .inpcrd file.
# Gets the starting position for every atom in the system and
# initializes the simulation
sim.context.setPositions(inpcrd.getPositions(asNumpy=True))
Next, add this section immediately before declaring the checkpoint file to use going forward.
## Open checkpoint file
with open("restart.chk",'rb') as f:
sim.context.loadCheckpoint(f.read())
You will also want to remove the line that minimizes the system, as you no
longer want this to happen.
Afterwards, the rest of the script is the same.
You may notice that the “restart.chk” file is differently named than the normal
checkpoint file we declared before.
You can technically use the same checkpoint file to restart and immediately
begin overwriting, but this has its own risks in case something goes wrong.
By making a copy of the checkpoint and restarting from the copy, you have a
separate backup that will allow for mistakes without losing data
(Author speaks from painful experience here!)
At this point, you should be able to set up an OpenMM simulation from start to finish, including resuming a stopped simulation.
Key Points
OpenMM in Python can make use of GPUs to perform molecular dynamics simulations.
If you continue a simulation from a checkpoint file, it is highly advised that you use a different filename for future checkpoint files.
MDAnalysis
Overview
Teaching: 25 min
Exercises: 5 minQuestions
How are structure and trajectory files read with Python?
What is an atom group?
How do I apply analyses to residues, instead of individual atoms?
How can tabular data files and plots be generated with Python?
Objectives
Load structures and trajectories into Python.
Use AtomGroups to make selections of atoms.
Construct an appropriate reference for analysis.
Understand how RMSF information is calculated in MDA.
List which force fields have hydrogen bond donor and acceptor pairs defined by default.
Highlights
- The Universe System
- Memory with MDAnalysis
- Loading Different File Types
- Load a Tinker XYZ
- Solution for Loading a Tinker XYZ
- Atom Groups
- Setting Up Analysis
- Writing Out Files
pandasandmatplotlib
There is a real MDAnalysis tutorial. This is more of “highlights” lesson.
The Universe System
Topologies and coordinates are loaded in through universes in MDAnalysis (MDA). It can read in and write many of the common structure formats, including those used with AMBER, CHARMM, GAMESS, GROMACS, LAMMPS, and TINKER.
The only necessary argument for creating a universe is a topology-based
filename.
import MDAnalysis as mda
system = mda.Universe(filename, in_memory=True)
Memory with MDAnalysis
The above example includes the keyword argument
in_memory=True. Trajectory information can be a massive drain on compute resources, so by default, this is set toFalse, instead only loading 1 frame at a time. Thus, almost every analysis operation relies on usingforloops to iterate through every frame. For relatively small trajectories, however, (like QM/MM optimizations that are often less than 10 frames), it can be easier to just load in everything at once.
To change the loaded frame, you need to specify the frame number to change to
(again, keeping in mind that they start at zero).
You can use -1 to specify the final frame.
u.trajectory[-1]
You can verify that the timestep has been changed by printing it.
> u.trajectory.ts
< Timestep 99 with unit cell dimensions [76.5608 70.27321 80.72006 90. 90. 90. ] >
Periodic boundary conditions (PBC) are assumed by default. To turn off PBC, you would need to use an on-the-fly transformation (not covered here).
Loading Different File Types
Most of the file types can be read using their commonly used extensions,
however, there are certainly times where specifying the file format is
necessary.
For instance, XYZ files and TINKER XYZ files are formatted differently, but
both may use the XYZ extension.
Thus, it is important to specify the TINKER format, because the file would only
be read correctly if the extension was .txyz.
## Read in a TINKER XYZ
system = mda.Universe("my_tinker_file.xyz", format='TXYZ')
Trajectories can be loaded in a chain format, meaning that you can “stack” them after loading an initial topology file.
## Read in an AMBER topology and trajectory
system = mda.Universe("protein.prmtop", "protein_1.nc", "protein_2.nc",
"protein_3.nc", "protein_4.nc", "protein_5.nc")
One of the keyword arguments you can feed the Universe command is dt to
specify the timestep.
The dt value should be the time in picoseconds between two frames.
Load a Tinker XYZ
Create a universe where you use a PDB file for the topology and a TINKER XYZ for the trajectory. Then, check the type and ensure it is a universe.
Solution for Loading a Tinker XYZ
You can either define variables with the file names, or you can use them directly in the command associated with them. It is important to note that the PDB coordinates are read in this scenario.
import MDAnalysis as mda ## in_PDB is the original PDB, in_txyz is the converted TINKER XYZ in_pdb = "my_original.pdb" in_txyz = "my_original.xyz" ## Read in the PDB as topology and TXYZ as "trajectory" system = mda.Universe(in_pdb, in_txyz, format='TXYZ', in_memory=True) ## Check the type type(system)
Atom Groups
One of the best aspects of MDA is its atom selection language.
You can store specific atoms selected from a universe as new variables.
The command to do this is: mda.Universe.select_atoms("").
A short example for selecting all the alpha carbons (CA) in a protein is below.
import MDAnalysis as mda
u = mda.Universe("5Y2S_wat.prmtop", "5Y2S_wat.dcd")
C_alphas = u.select_atoms("protein and name CA")
C_alphas
<AtomGroup with 254 atoms>
You can also make selections with slices of atoms. These selections use the Python indexing system, not the numbering in the file. Thus, the count starts at zero (because Python starts at 0), and it does not include the final index.
first_ten = u.atoms[0:10]
print(first_ten)
<AtomGroup [<Atom 1: N of type N3 of resname HIE, resid 1 and segid SYSTEM>,
<Atom 2: H1 of type H of resname HIE, resid 1 and segid SYSTEM>,
...,
<Atom 9: HB3 of type HC of resname HIE, resid 1 and segid SYSTEM>,
<Atom 10: CG of type CC of resname HIE, resid 1 and segid SYSTEM>]>
To further emphasize this point, the 0 index is shown as Atom 1, and the
10 index, which would then be listed as Atom 11 is not included.
One way to select atoms by the atom indices within the file is with bynum.
res_15 = u.atoms.select_atoms("bynum 226:247")
print(res_15)
<AtomGroup [<Atom 226: N of type N of resname LYS, resid 15 and segid SYSTEM>,
<Atom 227: H of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 228: CA of type CX of resname LYS, resid 15 and segid SYSTEM>,
...,
<Atom 245: HZ3 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 246: C of type C of resname LYS, resid 15 and segid SYSTEM>,
<Atom 247: O of type O of resname LYS, resid 15 and segid SYSTEM>]>
The above example selected all the atoms of a residue by number, but the
resnum keyword can do similarly.
You can use list(group_name[:]) to print everything in that group.
LYS_15 = u.select_atoms("resnum 15")
list(LYS_15[:])
[<Atom 226: N of type N of resname LYS, resid 15 and segid SYSTEM>,
<Atom 227: H of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 228: CA of type CX of resname LYS, resid 15 and segid SYSTEM>,
<Atom 229: HA of type H1 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 230: CB of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 231: HB2 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 232: HB3 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 233: CG of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 234: HG2 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 235: HG3 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 236: CD of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 237: HD2 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 238: HD3 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 239: CE of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 240: HE2 of type HP of resname LYS, resid 15 and segid SYSTEM>,
<Atom 241: HE3 of type HP of resname LYS, resid 15 and segid SYSTEM>,
<Atom 242: NZ of type N3 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 243: HZ1 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 244: HZ2 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 245: HZ3 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 246: C of type C of resname LYS, resid 15 and segid SYSTEM>,
<Atom 247: O of type O of resname LYS, resid 15 and segid SYSTEM>]
For many systems, "resnum 15" and "resid 15" should both give the same
result.
MDA also has syntax for selections based on masks, such as atoms or distance. Distance masks select atoms within in a certain distance cutoff (in angstroms).
This example would select any atoms within 5 Å of the ZN6 residue.
ZN_WAT = u.select_atoms("around 5 resname ZN6")
> ZN_WAT
<AtomGroup with 46 atoms>
You can extend this listing to include the entire residues that contain those atoms.
ZN_WAT = ZN_WAT.residues.atoms
> ZN_WAT
<AtomGroup with 150 atoms>
In the example, there were 46 atoms within a 5 A radius, but 150 total atoms for all the residues with atoms within that radius.
AtomGroups can be combined into larger groups, as well.
If order is important, use +. This may keep duplicates.
If it is important to not repeat atoms, then | makes a union.
You can force a group to be sorted and without duplicates after-the-fact with
unique.
## Just adds them and keeps them unordered
res_300 = u.select_atoms("resnum 300")
ZN_WAT = u.select_atoms("around 5 resname ZN6")
ZN_coord_and_300 = res_300 + ZN_WAT + res_300
> ZN_coord_and_300
<AtomGroup with 52 atoms>
## Makes a union without duplicates
res_300 = u.select_atoms("resnum 300")
ZN_WAT = u.select_atoms("around 5 resname ZN6")
ZN_coord_and_300 = res_300 | ZN_WAT | res_300
> ZN_coord_and_300
<AtomGroup with 49 atoms>
res_300 = u.select_atoms("resnum 300")
ZN_WAT = u.select_atoms("around 5 resname ZN6")
ZN_coord_and_300 = res_300 + ZN_WAT + res_300
ZN_coord_and_300_u = ZN_coord_and_300.unique
> ZN_coord_and_300_u
<AtomGroup with 52 atoms>
Setting Up Analysis
MDA keeps a lot of functions in sub-modules that also must be loaded in, which is one way to keep the package loading relevant.
import MDAnalysis as mda
from MDAnalysis.analysis import align, rms, hbonds
The bulk of analysis scripts using MDAnalysis will be setting up precisely what you’re trying to analyze or compare.
There is currently a GitHub issue about potentially creating command-line tools for MDA.
Alignments
It can be useful to align different structures (like those with mutations).
The mda.analysis.align.AlignTraj(moving, reference) command can align an
entire trajectory to a specified reference structure.
The reference group is the second **kwarg.
However, because of the assumption that PBC are turned on for the data, this
should not be necessary for general analyses.
After setting up the information to run the alignment, use .run() to
perform the action.
moving = u.select_atoms("protein")
reference = ref.select_atoms("protein")
alignment = mda.analysis.align.AlignTraj(moving.universe, reference.universe)
alignment.run()
<MDAnalysis.analysis.align.AlignTraj object at 0x8261b0ac8>
The alignment is now stored as alignment (though, like all things in Python,
you can name your own variables as long as you’re consistent).
RMSD
RMSD can compare the root mean square deviation between two similarly shaped
groups.
When using data from periodic boundary simulations, use whole-residues for
comparison.
The actual command to set up an analysis is
mda.analysis.rms.RMSD(group, reference_group). This is then run with .run()
## Trajectory
system = mda.Universe("system.topology", "system.traj")
## Select residues/atoms of interest to calculate RMSD (inclusive)
ROI = 'resid 1:261'
## Reference (Crystal PDB)
ref = mda.Universe("crystal.pdb")
## Select atoms for RMSD
resnames = system.select_atoms(ROI)
resnames_ref = ref.select_atoms(ROI)
## Get the RMSDs
rmsd = mda.analysis.rms.RMSD(resnames, resnames_ref)
rmsd.run()
## Create a Pandas dataframe with info
rmsd_df = pd.DataFrame(columns = ['#Frame', 'RMSD'])
for i in range(len(rmsd.rmsd)):
rmsd_df = rmsd_df.append({
## First index is frame, then info is Frame, Time, RMSD aka [0, 1, 2]
"#Frame" : rmsd.rmsd[i][0],
"RMSD" : rmsd.rmsd[i][2]
}, ignore_index=True)
## Round to 4 digits
rmsd_df = rmsd_df.round(4)
rmsd_df.to_csv("rmsd.dat", sep='\t', index=False, encoding='utf8', header=True)
After .run(), the information can be saved as a pandas dataframe
(assuming you loaded it with import pandas as pd!!!)
pandas is a really handy package for working with tabular data and saving CSV
files.
The final line with to_csv saves the RMSD information to a file.
RMSF
Root mean square fluctuation (RMSF) information can also be calculated.
Since this is performed by-atom, the residue weights need to be
applied to the by-atom fluctuations in order to get the RMSF per residue.
The basic RMSF analysis is called with mda.analysis.rms.RMSF(resnames) and
then run with .run().
The weighting part first gets the residue weights for every atom in the
specified atom group before using those weights through accumulate to apply
the average to the residue.
## Run for individual residues
rmsf = mda.analysis.rms.RMSF(resnames)
rmsf.run()
## Apply weights to the by-atom fluctuations
weight_rmsf = []
for atom in range(len(rmsf.rmsf)):
## weight_rmsf = fluctuation * atom_mass / residue_mass
weight_rmsf.append( (rmsf.rmsf[atom] *
( res_weights[atom] / rmsf.atomgroup[atom].residue.mass)) )
## Combine into by-residue value
byres_rmsf = resnames.accumulate(weight_rmsf, compound='residues')
## Create a Pandas dataframe with info
rmsf_df = pd.DataFrame(columns = ['#Res', 'AtomicFlx'])
for i in range(len(byres_rmsf)):
rmsf_df = rmsf_df.append({
"#Res" : resnames.residues[i].resnum,
"AtomicFlx" : byres_rmsf[i]
}, ignore_index=True)
## Round to 4 digits
rmsf_df = rmsf_df.round(4)
rmsf_df.to_csv("rmsf.dat", sep='\t', index=False, encoding='utf8', header=True)
Like before, a pandas dataframe is created.
Hydrogen Bond Analysis
MDA’s search criteria for hydrogen bond analysis (HBA) does not have information for AMBER, but it can be written explicitly as a new HBA class. CHARMM and GLYCAM are the two force fields currently written into MDA for HBA. When making the class that includes AMBER, it is safer to define the new HBA command as a unique class, instead of redefining the MDA package’s class.
class HydrogenBondAnalysis_AMBER(mda.analysis.hbonds.HydrogenBondAnalysis):
# use tuple(set()) here so that one can just copy&paste names from the
# table; set() takes care for removing duplicates. At the end the
# DEFAULT_DONORS and DEFAULT_ACCEPTORS should simply be tuples.
#
#: default heavy atom names whose hydrogens are treated as *donors*
#: (see :ref:`Default atom names for hydrogen bonding analysis`);
#: use the keyword `donors` to add a list of additional donor names.
DEFAULT_DONORS = {
'AMBER': tuple(set([
'N','OG','OG1','OE2','OH','NH1','NH2','NZ','OH','N2','ND1','NE', \
'NE1','NE2','N2','N3','N4','N6','O5\'', 'OD2', ])),
'CHARMM27': tuple(set([
'N', 'OH2', 'OW', 'NE', 'NH1', 'NH2', 'ND2', 'SG', 'NE2', 'ND1', \
'NZ', 'OG', 'OG1', 'NE1', 'OH'])),
'GLYCAM06': tuple(set(['N', 'NT', 'N3', 'OH', 'OW'])),
'other': tuple(set([]))}
#
#: default atom names that are treated as hydrogen *acceptors*
#: (see :ref:`Default atom names for hydrogen bonding analysis`);
#: use the keyword `acceptors` to add a list of additional acceptor names.
DEFAULT_ACCEPTORS = {
'AMBER': tuple(set([
'O','OD1','OD2','OE1','OE2','N','ND1','NE2','NZ','OG','OG1','O1',\
'O2','O4','O6','N1','N3','N6','N7','O1P','O2P','O3\'','O4\'','OH'])),
'CHARMM27': tuple(set([
'O', 'OC1', 'OC2', 'OH2', 'OW', 'OD1', 'OD2', 'SG', 'OE1', 'OE1', \
'OE2', 'ND1', 'NE2', 'SD', 'OG', 'OG1', 'OH'])),
'GLYCAM06': tuple(set(['N', 'NT', 'O', 'O2', 'OH', 'OS', 'OW', 'OY', 'SM'])),
'other': tuple(set([]))}
After it is defined, it can be run like the other analyses, specifying the
default distance and angle information. This is one analysis where pbc needs
to explicity be set to True for simulations with periodic boundary conditions.
hbond = HydrogenBondAnalysis_AMBER(system, ROI, ROI, distance=3.0, angle=135.0, \
forcefield='AMBER', pbc=True)
hbond.run()
## Save by frequency
new_hbond_df = pd.DataFrame(hbond.count_by_type())
new_hbond_df = new_hbond_df.sort_values(by=['frequency'], ascending=False)
new_hbond_df.to_csv("hbond.dat", sep='\t', index=False, encoding='utf8', header=True)
The HBA class was designed to be a little friendlier with pandas, which
is why there are not column names that have to be assigned.
Writing Out Files
MDA can write out several formats, for single frames or many frames.
By default, it will write out the current frame that is loaded for single
frame usage.
You can print the information for the loaded frame through something like
u.universe.trajectory.ts, where u is a loaded universe.
u.atoms.write("5Y2S_from_MDA.pdb")
This can be written more Pythonically, while explicitly specifying the frame.
with mda.Writer(outfile, multiframe=False) as pdb:
system.trajectory[-1]
pdb.write(system)
pandas and matplotlib
Now that we’ve talked about analysis, we can touch a little bit more about
using pandas and matplotlib.
A lot of the MDA analysis commands wrote very cleanly to pandas dataframes.
These dataframes can often be easily plotted using matplotlib, a very
broadly used Python module for data visualization.
This is a minimal working example for reading in a data file as a dataframe
and plotting #Frame on the x-axis and RMSD on the y-axis.
The labels are then updated to reflect time and RMSD.
The plot is then saved as a .png using plt.savefig at 300 DPI.
import pandas as pd
import matplotlib.pyplot as plt
rmsd_in = '5Y2S_rmsd.dat'
rmsd_plot = '5Y2S_RMSD.png'
## Read the file as a pandas dataframe
rmsd_df = pd.read_csv(rmsd_in, sep='\t')
## Plot x and y
plt.plot(rmsd_df['#Frame'], rmsd_df['RMSD'])
## Update axes labels
plt.xlabel('Time (ps)')
plt.ylabel('RMSD ($\AA$)')
## Save the figure
plt.savefig(rmsd_plot, dpi=300)
The reading of a dataframe can be skipped using plt.plotfile.
import pandas as pd
import matplotlib.pyplot as plt
rmsd_in = '5Y2S_rmsd.dat'
rmsd_plot = '5Y2S_RMSD.png'
## Plot the pandas dataframe while defining the axis labels with `names`
plt.plotfile(rmsd_in, cols=(0,1), delimiter='\t', \
names=('Time (ps)','RMSD ($\AA$)'))
## Save the figure
plt.savefig(rmsd_plot, dpi=300)
Both of these produce this graph.
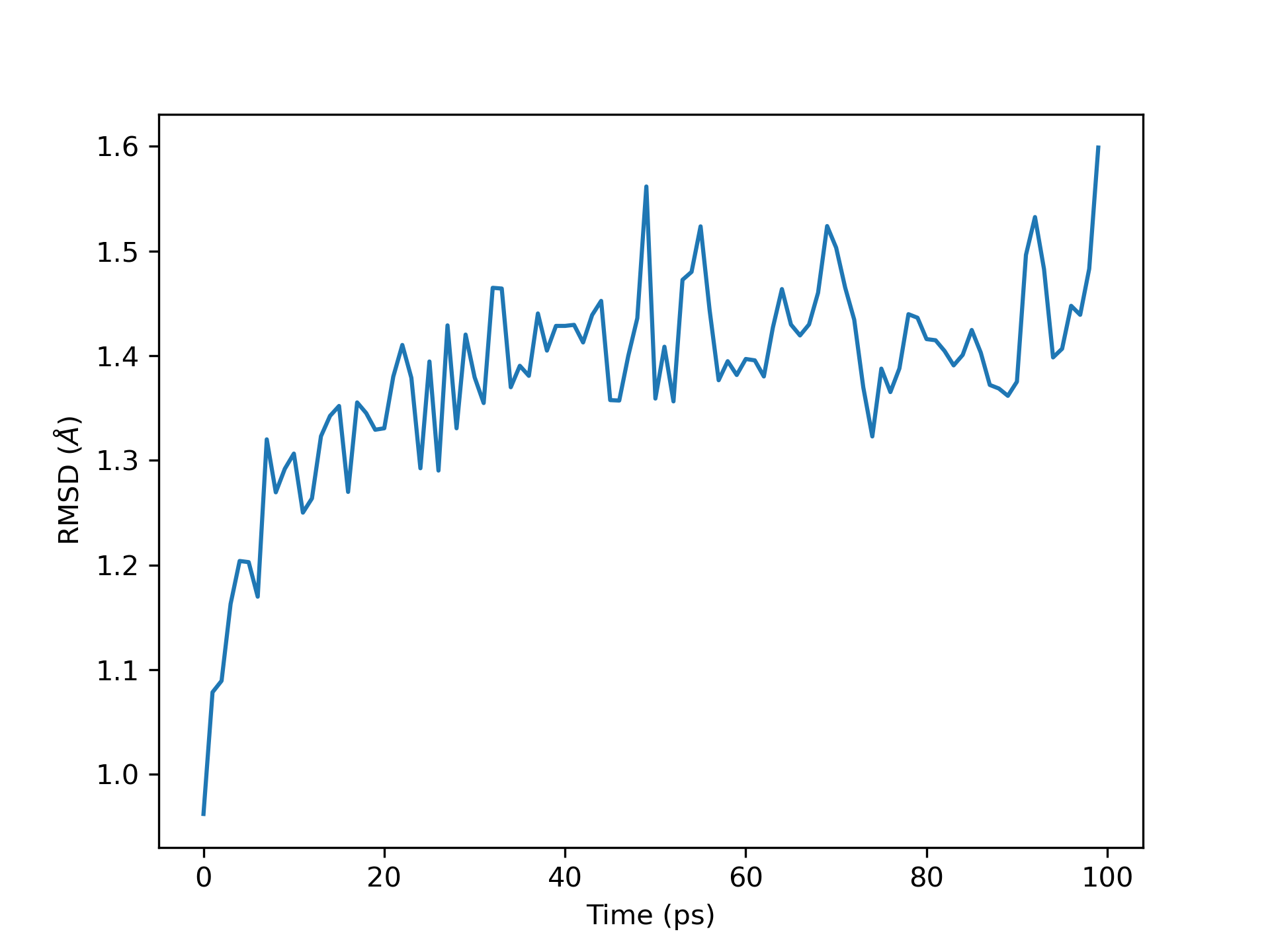
RMSD plot for 1 ns of simulation.
Key Points
MDA uses universes to store topology and coordinate information.
The
XYZextension is used for XYZs,TXYZis used for TINKER XYZs.Periodic boundary conditions are turned on by default.
Root mean square deviation (RMSD) must be calculated against a reference structure.
Root mean square fluctuation (RMSF) is calculated by-atom originally, and the residue weights must be applied to get the mass-weighted RMSF.
CHARMM27 and GLYCAM06 are the pre-defined force fields for hydrogen bond analysis. New force fields can be added by defining them through the
HydrogenBondAnalysisclass.