MDAnalysis
Overview
Teaching: 25 min
Exercises: 5 minQuestions
How are structure and trajectory files read with Python?
What is an atom group?
How do I apply analyses to residues, instead of individual atoms?
How can tabular data files and plots be generated with Python?
Objectives
Load structures and trajectories into Python.
Use AtomGroups to make selections of atoms.
Construct an appropriate reference for analysis.
Understand how RMSF information is calculated in MDA.
List which force fields have hydrogen bond donor and acceptor pairs defined by default.
Highlights
- The Universe System
- Memory with MDAnalysis
- Loading Different File Types
- Load a Tinker XYZ
- Solution for Loading a Tinker XYZ
- Atom Groups
- Setting Up Analysis
- Writing Out Files
pandasandmatplotlib
There is a real MDAnalysis tutorial. This is more of “highlights” lesson.
The Universe System
Topologies and coordinates are loaded in through universes in MDAnalysis (MDA). It can read in and write many of the common structure formats, including those used with AMBER, CHARMM, GAMESS, GROMACS, LAMMPS, and TINKER.
The only necessary argument for creating a universe is a topology-based
filename.
import MDAnalysis as mda
system = mda.Universe(filename, in_memory=True)
Memory with MDAnalysis
The above example includes the keyword argument
in_memory=True. Trajectory information can be a massive drain on compute resources, so by default, this is set toFalse, instead only loading 1 frame at a time. Thus, almost every analysis operation relies on usingforloops to iterate through every frame. For relatively small trajectories, however, (like QM/MM optimizations that are often less than 10 frames), it can be easier to just load in everything at once.
To change the loaded frame, you need to specify the frame number to change to
(again, keeping in mind that they start at zero).
You can use -1 to specify the final frame.
u.trajectory[-1]
You can verify that the timestep has been changed by printing it.
> u.trajectory.ts
< Timestep 99 with unit cell dimensions [76.5608 70.27321 80.72006 90. 90. 90. ] >
Periodic boundary conditions (PBC) are assumed by default. To turn off PBC, you would need to use an on-the-fly transformation (not covered here).
Loading Different File Types
Most of the file types can be read using their commonly used extensions,
however, there are certainly times where specifying the file format is
necessary.
For instance, XYZ files and TINKER XYZ files are formatted differently, but
both may use the XYZ extension.
Thus, it is important to specify the TINKER format, because the file would only
be read correctly if the extension was .txyz.
## Read in a TINKER XYZ
system = mda.Universe("my_tinker_file.xyz", format='TXYZ')
Trajectories can be loaded in a chain format, meaning that you can “stack” them after loading an initial topology file.
## Read in an AMBER topology and trajectory
system = mda.Universe("protein.prmtop", "protein_1.nc", "protein_2.nc",
"protein_3.nc", "protein_4.nc", "protein_5.nc")
One of the keyword arguments you can feed the Universe command is dt to
specify the timestep.
The dt value should be the time in picoseconds between two frames.
Load a Tinker XYZ
Create a universe where you use a PDB file for the topology and a TINKER XYZ for the trajectory. Then, check the type and ensure it is a universe.
Solution for Loading a Tinker XYZ
You can either define variables with the file names, or you can use them directly in the command associated with them. It is important to note that the PDB coordinates are read in this scenario.
import MDAnalysis as mda ## in_PDB is the original PDB, in_txyz is the converted TINKER XYZ in_pdb = "my_original.pdb" in_txyz = "my_original.xyz" ## Read in the PDB as topology and TXYZ as "trajectory" system = mda.Universe(in_pdb, in_txyz, format='TXYZ', in_memory=True) ## Check the type type(system)
Atom Groups
One of the best aspects of MDA is its atom selection language.
You can store specific atoms selected from a universe as new variables.
The command to do this is: mda.Universe.select_atoms("").
A short example for selecting all the alpha carbons (CA) in a protein is below.
import MDAnalysis as mda
u = mda.Universe("5Y2S_wat.prmtop", "5Y2S_wat.dcd")
C_alphas = u.select_atoms("protein and name CA")
C_alphas
<AtomGroup with 254 atoms>
You can also make selections with slices of atoms. These selections use the Python indexing system, not the numbering in the file. Thus, the count starts at zero (because Python starts at 0), and it does not include the final index.
first_ten = u.atoms[0:10]
print(first_ten)
<AtomGroup [<Atom 1: N of type N3 of resname HIE, resid 1 and segid SYSTEM>,
<Atom 2: H1 of type H of resname HIE, resid 1 and segid SYSTEM>,
...,
<Atom 9: HB3 of type HC of resname HIE, resid 1 and segid SYSTEM>,
<Atom 10: CG of type CC of resname HIE, resid 1 and segid SYSTEM>]>
To further emphasize this point, the 0 index is shown as Atom 1, and the
10 index, which would then be listed as Atom 11 is not included.
One way to select atoms by the atom indices within the file is with bynum.
res_15 = u.atoms.select_atoms("bynum 226:247")
print(res_15)
<AtomGroup [<Atom 226: N of type N of resname LYS, resid 15 and segid SYSTEM>,
<Atom 227: H of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 228: CA of type CX of resname LYS, resid 15 and segid SYSTEM>,
...,
<Atom 245: HZ3 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 246: C of type C of resname LYS, resid 15 and segid SYSTEM>,
<Atom 247: O of type O of resname LYS, resid 15 and segid SYSTEM>]>
The above example selected all the atoms of a residue by number, but the
resnum keyword can do similarly.
You can use list(group_name[:]) to print everything in that group.
LYS_15 = u.select_atoms("resnum 15")
list(LYS_15[:])
[<Atom 226: N of type N of resname LYS, resid 15 and segid SYSTEM>,
<Atom 227: H of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 228: CA of type CX of resname LYS, resid 15 and segid SYSTEM>,
<Atom 229: HA of type H1 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 230: CB of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 231: HB2 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 232: HB3 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 233: CG of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 234: HG2 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 235: HG3 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 236: CD of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 237: HD2 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 238: HD3 of type HC of resname LYS, resid 15 and segid SYSTEM>,
<Atom 239: CE of type C8 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 240: HE2 of type HP of resname LYS, resid 15 and segid SYSTEM>,
<Atom 241: HE3 of type HP of resname LYS, resid 15 and segid SYSTEM>,
<Atom 242: NZ of type N3 of resname LYS, resid 15 and segid SYSTEM>,
<Atom 243: HZ1 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 244: HZ2 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 245: HZ3 of type H of resname LYS, resid 15 and segid SYSTEM>,
<Atom 246: C of type C of resname LYS, resid 15 and segid SYSTEM>,
<Atom 247: O of type O of resname LYS, resid 15 and segid SYSTEM>]
For many systems, "resnum 15" and "resid 15" should both give the same
result.
MDA also has syntax for selections based on masks, such as atoms or distance. Distance masks select atoms within in a certain distance cutoff (in angstroms).
This example would select any atoms within 5 Å of the ZN6 residue.
ZN_WAT = u.select_atoms("around 5 resname ZN6")
> ZN_WAT
<AtomGroup with 46 atoms>
You can extend this listing to include the entire residues that contain those atoms.
ZN_WAT = ZN_WAT.residues.atoms
> ZN_WAT
<AtomGroup with 150 atoms>
In the example, there were 46 atoms within a 5 A radius, but 150 total atoms for all the residues with atoms within that radius.
AtomGroups can be combined into larger groups, as well.
If order is important, use +. This may keep duplicates.
If it is important to not repeat atoms, then | makes a union.
You can force a group to be sorted and without duplicates after-the-fact with
unique.
## Just adds them and keeps them unordered
res_300 = u.select_atoms("resnum 300")
ZN_WAT = u.select_atoms("around 5 resname ZN6")
ZN_coord_and_300 = res_300 + ZN_WAT + res_300
> ZN_coord_and_300
<AtomGroup with 52 atoms>
## Makes a union without duplicates
res_300 = u.select_atoms("resnum 300")
ZN_WAT = u.select_atoms("around 5 resname ZN6")
ZN_coord_and_300 = res_300 | ZN_WAT | res_300
> ZN_coord_and_300
<AtomGroup with 49 atoms>
res_300 = u.select_atoms("resnum 300")
ZN_WAT = u.select_atoms("around 5 resname ZN6")
ZN_coord_and_300 = res_300 + ZN_WAT + res_300
ZN_coord_and_300_u = ZN_coord_and_300.unique
> ZN_coord_and_300_u
<AtomGroup with 52 atoms>
Setting Up Analysis
MDA keeps a lot of functions in sub-modules that also must be loaded in, which is one way to keep the package loading relevant.
import MDAnalysis as mda
from MDAnalysis.analysis import align, rms, hbonds
The bulk of analysis scripts using MDAnalysis will be setting up precisely what you’re trying to analyze or compare.
There is currently a GitHub issue about potentially creating command-line tools for MDA.
Alignments
It can be useful to align different structures (like those with mutations).
The mda.analysis.align.AlignTraj(moving, reference) command can align an
entire trajectory to a specified reference structure.
The reference group is the second **kwarg.
However, because of the assumption that PBC are turned on for the data, this
should not be necessary for general analyses.
After setting up the information to run the alignment, use .run() to
perform the action.
moving = u.select_atoms("protein")
reference = ref.select_atoms("protein")
alignment = mda.analysis.align.AlignTraj(moving.universe, reference.universe)
alignment.run()
<MDAnalysis.analysis.align.AlignTraj object at 0x8261b0ac8>
The alignment is now stored as alignment (though, like all things in Python,
you can name your own variables as long as you’re consistent).
RMSD
RMSD can compare the root mean square deviation between two similarly shaped
groups.
When using data from periodic boundary simulations, use whole-residues for
comparison.
The actual command to set up an analysis is
mda.analysis.rms.RMSD(group, reference_group). This is then run with .run()
## Trajectory
system = mda.Universe("system.topology", "system.traj")
## Select residues/atoms of interest to calculate RMSD (inclusive)
ROI = 'resid 1:261'
## Reference (Crystal PDB)
ref = mda.Universe("crystal.pdb")
## Select atoms for RMSD
resnames = system.select_atoms(ROI)
resnames_ref = ref.select_atoms(ROI)
## Get the RMSDs
rmsd = mda.analysis.rms.RMSD(resnames, resnames_ref)
rmsd.run()
## Create a Pandas dataframe with info
rmsd_df = pd.DataFrame(columns = ['#Frame', 'RMSD'])
for i in range(len(rmsd.rmsd)):
rmsd_df = rmsd_df.append({
## First index is frame, then info is Frame, Time, RMSD aka [0, 1, 2]
"#Frame" : rmsd.rmsd[i][0],
"RMSD" : rmsd.rmsd[i][2]
}, ignore_index=True)
## Round to 4 digits
rmsd_df = rmsd_df.round(4)
rmsd_df.to_csv("rmsd.dat", sep='\t', index=False, encoding='utf8', header=True)
After .run(), the information can be saved as a pandas dataframe
(assuming you loaded it with import pandas as pd!!!)
pandas is a really handy package for working with tabular data and saving CSV
files.
The final line with to_csv saves the RMSD information to a file.
RMSF
Root mean square fluctuation (RMSF) information can also be calculated.
Since this is performed by-atom, the residue weights need to be
applied to the by-atom fluctuations in order to get the RMSF per residue.
The basic RMSF analysis is called with mda.analysis.rms.RMSF(resnames) and
then run with .run().
The weighting part first gets the residue weights for every atom in the
specified atom group before using those weights through accumulate to apply
the average to the residue.
## Run for individual residues
rmsf = mda.analysis.rms.RMSF(resnames)
rmsf.run()
## Apply weights to the by-atom fluctuations
weight_rmsf = []
for atom in range(len(rmsf.rmsf)):
## weight_rmsf = fluctuation * atom_mass / residue_mass
weight_rmsf.append( (rmsf.rmsf[atom] *
( res_weights[atom] / rmsf.atomgroup[atom].residue.mass)) )
## Combine into by-residue value
byres_rmsf = resnames.accumulate(weight_rmsf, compound='residues')
## Create a Pandas dataframe with info
rmsf_df = pd.DataFrame(columns = ['#Res', 'AtomicFlx'])
for i in range(len(byres_rmsf)):
rmsf_df = rmsf_df.append({
"#Res" : resnames.residues[i].resnum,
"AtomicFlx" : byres_rmsf[i]
}, ignore_index=True)
## Round to 4 digits
rmsf_df = rmsf_df.round(4)
rmsf_df.to_csv("rmsf.dat", sep='\t', index=False, encoding='utf8', header=True)
Like before, a pandas dataframe is created.
Hydrogen Bond Analysis
MDA’s search criteria for hydrogen bond analysis (HBA) does not have information for AMBER, but it can be written explicitly as a new HBA class. CHARMM and GLYCAM are the two force fields currently written into MDA for HBA. When making the class that includes AMBER, it is safer to define the new HBA command as a unique class, instead of redefining the MDA package’s class.
class HydrogenBondAnalysis_AMBER(mda.analysis.hbonds.HydrogenBondAnalysis):
# use tuple(set()) here so that one can just copy&paste names from the
# table; set() takes care for removing duplicates. At the end the
# DEFAULT_DONORS and DEFAULT_ACCEPTORS should simply be tuples.
#
#: default heavy atom names whose hydrogens are treated as *donors*
#: (see :ref:`Default atom names for hydrogen bonding analysis`);
#: use the keyword `donors` to add a list of additional donor names.
DEFAULT_DONORS = {
'AMBER': tuple(set([
'N','OG','OG1','OE2','OH','NH1','NH2','NZ','OH','N2','ND1','NE', \
'NE1','NE2','N2','N3','N4','N6','O5\'', 'OD2', ])),
'CHARMM27': tuple(set([
'N', 'OH2', 'OW', 'NE', 'NH1', 'NH2', 'ND2', 'SG', 'NE2', 'ND1', \
'NZ', 'OG', 'OG1', 'NE1', 'OH'])),
'GLYCAM06': tuple(set(['N', 'NT', 'N3', 'OH', 'OW'])),
'other': tuple(set([]))}
#
#: default atom names that are treated as hydrogen *acceptors*
#: (see :ref:`Default atom names for hydrogen bonding analysis`);
#: use the keyword `acceptors` to add a list of additional acceptor names.
DEFAULT_ACCEPTORS = {
'AMBER': tuple(set([
'O','OD1','OD2','OE1','OE2','N','ND1','NE2','NZ','OG','OG1','O1',\
'O2','O4','O6','N1','N3','N6','N7','O1P','O2P','O3\'','O4\'','OH'])),
'CHARMM27': tuple(set([
'O', 'OC1', 'OC2', 'OH2', 'OW', 'OD1', 'OD2', 'SG', 'OE1', 'OE1', \
'OE2', 'ND1', 'NE2', 'SD', 'OG', 'OG1', 'OH'])),
'GLYCAM06': tuple(set(['N', 'NT', 'O', 'O2', 'OH', 'OS', 'OW', 'OY', 'SM'])),
'other': tuple(set([]))}
After it is defined, it can be run like the other analyses, specifying the
default distance and angle information. This is one analysis where pbc needs
to explicity be set to True for simulations with periodic boundary conditions.
hbond = HydrogenBondAnalysis_AMBER(system, ROI, ROI, distance=3.0, angle=135.0, \
forcefield='AMBER', pbc=True)
hbond.run()
## Save by frequency
new_hbond_df = pd.DataFrame(hbond.count_by_type())
new_hbond_df = new_hbond_df.sort_values(by=['frequency'], ascending=False)
new_hbond_df.to_csv("hbond.dat", sep='\t', index=False, encoding='utf8', header=True)
The HBA class was designed to be a little friendlier with pandas, which
is why there are not column names that have to be assigned.
Writing Out Files
MDA can write out several formats, for single frames or many frames.
By default, it will write out the current frame that is loaded for single
frame usage.
You can print the information for the loaded frame through something like
u.universe.trajectory.ts, where u is a loaded universe.
u.atoms.write("5Y2S_from_MDA.pdb")
This can be written more Pythonically, while explicitly specifying the frame.
with mda.Writer(outfile, multiframe=False) as pdb:
system.trajectory[-1]
pdb.write(system)
pandas and matplotlib
Now that we’ve talked about analysis, we can touch a little bit more about
using pandas and matplotlib.
A lot of the MDA analysis commands wrote very cleanly to pandas dataframes.
These dataframes can often be easily plotted using matplotlib, a very
broadly used Python module for data visualization.
This is a minimal working example for reading in a data file as a dataframe
and plotting #Frame on the x-axis and RMSD on the y-axis.
The labels are then updated to reflect time and RMSD.
The plot is then saved as a .png using plt.savefig at 300 DPI.
import pandas as pd
import matplotlib.pyplot as plt
rmsd_in = '5Y2S_rmsd.dat'
rmsd_plot = '5Y2S_RMSD.png'
## Read the file as a pandas dataframe
rmsd_df = pd.read_csv(rmsd_in, sep='\t')
## Plot x and y
plt.plot(rmsd_df['#Frame'], rmsd_df['RMSD'])
## Update axes labels
plt.xlabel('Time (ps)')
plt.ylabel('RMSD ($\AA$)')
## Save the figure
plt.savefig(rmsd_plot, dpi=300)
The reading of a dataframe can be skipped using plt.plotfile.
import pandas as pd
import matplotlib.pyplot as plt
rmsd_in = '5Y2S_rmsd.dat'
rmsd_plot = '5Y2S_RMSD.png'
## Plot the pandas dataframe while defining the axis labels with `names`
plt.plotfile(rmsd_in, cols=(0,1), delimiter='\t', \
names=('Time (ps)','RMSD ($\AA$)'))
## Save the figure
plt.savefig(rmsd_plot, dpi=300)
Both of these produce this graph.
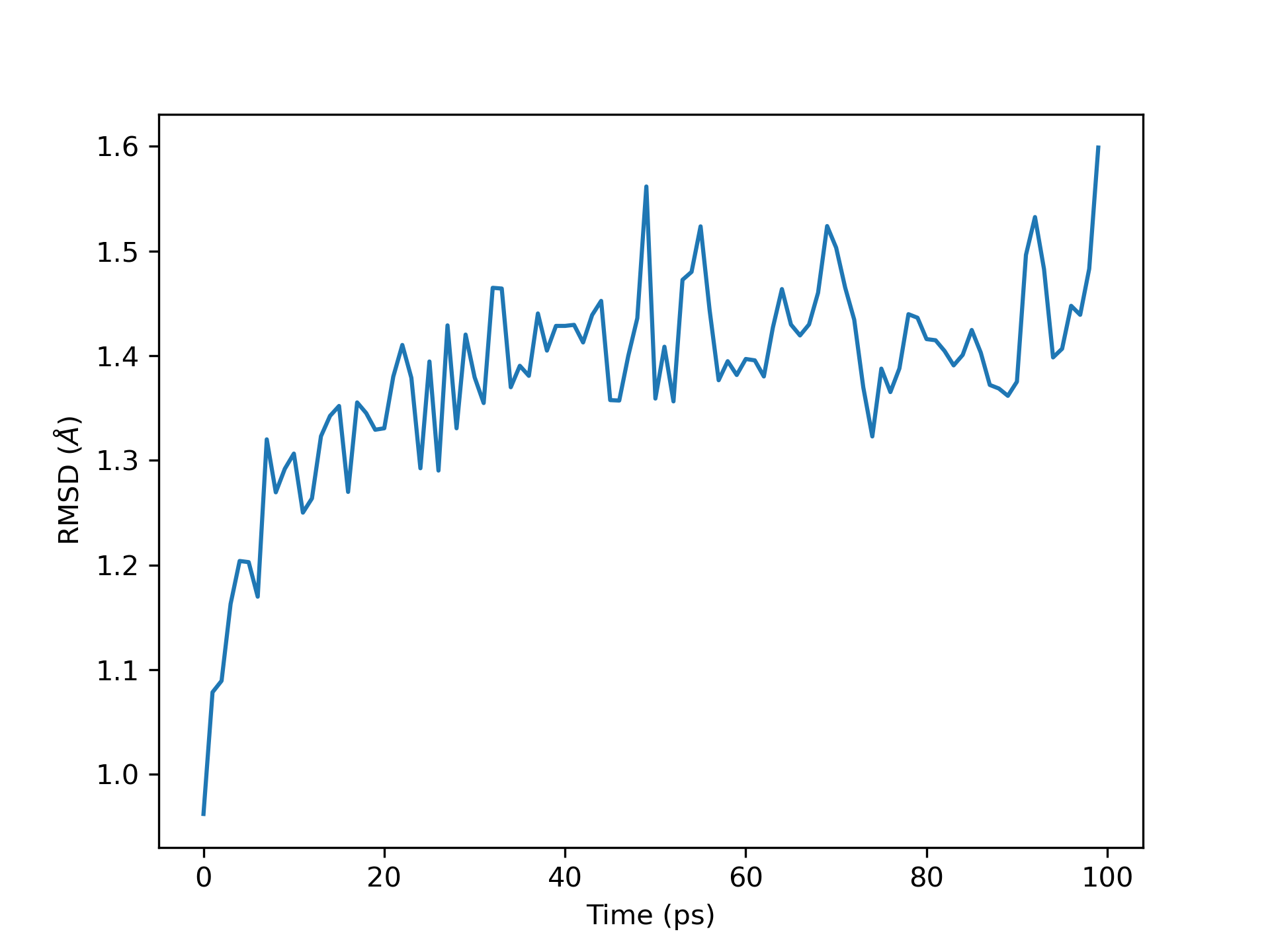
RMSD plot for 1 ns of simulation.
Key Points
MDA uses universes to store topology and coordinate information.
The
XYZextension is used for XYZs,TXYZis used for TINKER XYZs.Periodic boundary conditions are turned on by default.
Root mean square deviation (RMSD) must be calculated against a reference structure.
Root mean square fluctuation (RMSF) is calculated by-atom originally, and the residue weights must be applied to get the mass-weighted RMSF.
CHARMM27 and GLYCAM06 are the pre-defined force fields for hydrogen bond analysis. New force fields can be added by defining them through the
HydrogenBondAnalysisclass.