Introduction
Overview
Teaching: 15 min
Exercises: 0 minQuestions
What is LICHEM?
What is QM/MM? (FIXME)
Objectives
FIXME
LICHEM (Layered Interacting CHEmical Models) is an open-source package for interfacing between software for quantum mechanics (QM) and molecular mechanics (MM), enabling QM/MM calculations to make use of polarizable, frozen electron density, and point-charge force fields.
The LICHEM code is available on GitHub.
Citing LICHEM
Kratz, E.G.; Walker, A.R.; Lagardere, L; Lipparini, F; Piquemal, J.-P.; and Cisneros, G.A.; "LICHEM: A QM/MM Program for Simulations with Multipolar and Polarizable Force Fields", J. Comput. Chem., 37, 11, 1019, (2016). Gokcan H., Vazquez-Montelongo E.A., Cisneros G.A., “LICHEM 1.1: Recent Improvements and New Capabilities”, J. Chem. Theo. Comput., 15, 3056, (2019).
About QM/MM
Eventually, I will talk about the theory behind QM/MM here.
About the Pseudobond Approach
Eventually, I will talk about the pseudobond stuff here.
Selecting a QM Region
Eventually, I will talk about how to select a QM region here.
Key Points
FIXME
Structure Preparation
Overview
Teaching: 120 min
Exercises: 0 minQuestions
How do I make a structure biologically relevant?
Do I need to add hydrogens?
Where do parameters come from?
How do I add ions and solvate a system?
Objectives
Modify a crystal structure to reflect realistic protonation states.
Check a structure for clashes.
Create parameters for a small, organic molecule.
Generate a solvated and charge-neutral structure with LEaP.
Highlights
- Where are the files?
- PDB Structure Background
- Initial Processing
- Possible Error
- What in the world are all those
sedcommands for? - Hold up, why did we remove
GOLagain? - PROPKA
- MolProbity
- LEaP
- “Decently described” sounds sketchy…
- Using
tleap - Note about
solvatebox - Testimonial from a Grad Student: Read the Error Messages
Where are the files?
Check out the web
PDB Structure Background
The ATOM and HETATM records in a PDB file contain the relevant information
for getting started with computational simulations.
ATOM 1 N HIS 4 30.098 2.954 7.707 1.00 0.00 N
The records start with ATOM or HETATM.
Next is the atom index or atom ID number.
Each atom index is unique to that atom in a simulation, but before the structure
is fully prepared, these numbers will often be non-unique.
Next is the atom name. Atom names should be unique within a given residue.
So, for a methyl group with 3 hydrogens, those hydrogens would likely be labeled
as H1, H2, and H3.
After the atom name comes the residue name.
AMBER uses unique residue names for residues in different protonation states,
but not every program or force field follows this method.
Next is the residue number.
These should also be unique once the structure is fully prepared.
The following three numbers are the X, Y, and Z coordinates for the atom.
The next two numbers are the occupancy and the temperature factor, respectively.
RCSB has a good explanation of these in their
PDB-101
site, but they are not important right now.
Finally, the element symbol for the atom is listed.
Initial Processing
Download 5y2s.pdb from RCSB.
RCSB has a dropdown for “Display Files” and “Download Files.”
Choose the PDB format.
PDB files have a lot of crystallographic information that can confuse MD
programs. The important lines start with ATOM, HETATM, TER, and END.
The clean_pdb.sh file is a script that includes the following command to
extract these lines from the PDB file and save them in a new file.
grep -e '^ATOM\|^HETATM\|^TER\|^END' $thing > $thing_clean
To run the clean_pdb.sh script, use:
bash clean_pdb.sh
or
./clean_pdb.sh
Possible Error
If you try the second option (./clean_pdb.sh), you may see:
-bash: ./clean_pdb.sh: Permission deniedWhen scripts are initially written, they do not have the appropriate permissions to be run. This is actually an anti-malware strategy! You don’t just want commands to be able to run all willy nilly. That’s why you then need to use another command,
chmodto modify the file permissions and allow it to be executed (run).chmod u+x clean_pdb.sh
Running the clean_pdb.sh script creates the 5Y2S_clean.pdb file.
What in the world are all those
sedcommands for?Each of those
sedcommands is for in-place editing of the file. The first block removes “B chain” residues, the second block renames the “A chain” residues as the normal residue name, and the third block renamesHOHtoWATand removes theGOL, which is an inhibitor. PDBs often have “A chain” and “B chain” residues, which offer alternative occupancies for an atom. These occupancies are kind of like saying 65% of the day someone sits at a desk, but the other 35% of the day they lay in their bed. You would likely care most about the time spent at the desk, but there are also times that knowing they’re in bed is important. For the purpose of this tutorial, we’re taking all the A-chain residues and ignoring all the B-chain residues. Every system is different, and every structure is different, so it’s better to make a case-by-case decision on which of the chains to use for a residue.Hold up, why did we remove
GOLagain?There are a few common inhibitors for enzymes, and
GOL(glycerol) is often one of them. You can check for potential inhibitors by reading the paper published with the crystal structure (if one exists…). This is one case where reading the methods section is critically important.
PROPKA
The crystal structure doesn’t come with hydrogens. This is because they both rotate frequently, and they are very, very small compared to everything else, so they’re not actually captured. Because of that, hydrogens are placed based on an educated guess, but protonation states are pH dependent. Thus, we need to use something to predict the proper protonation states for each titratable residue. Here, we’ll use PROPKA.
Go to the PROPKA webserver and select PDB2PQR.
Upload the 5Y2S_clean.pdb file.
These options should be used with the webserver:
- pH 7.0
- Use PROPKA to assign protonation states at provided pH
- Forcefield: AMBER
- Choose output: AMBER
- Deselect Remove the waters from the output file
Once the job has completed, there are a number of files to download.
The relevant output files have the pqr, stdout.txt and stderr.txt
extensions.
The pqr file contains the adjusted residue names.
Not to skip ahead in the tutorial (that’s why these files are provided 😃),
but you can use MDAnalysis to convert the output PQR back to a PDB.
This is what the propka-reintegration.py script looks like.
import MDAnalysis as mda
propka_output="../2-propka_output/p09m0bn8cm.pqr"
pdb_out="5Y2S_ph7_protonated.pdb"
system = mda.Universe(propka_output, format="PQR", dt=1.0, in_memory=True)
## Remove the segment IDs (w/o this, `SYST` gets annoyingly printed by element)
for atom in system.atoms:
atom.segment.segid = ''
system.atoms.write(pdb_out)
A physical inspection of the .pqr file from PROPKA reveals these REMARK lines:
REMARK 5 WARNING: PDB2PQR was unable to assign charges
REMARK 5 to the following atoms (omitted below):
REMARK 5 2157 ZN in ZN 301
REMARK 5 2158 C in CO2 302
REMARK 5 2159 O1 in CO2 302
REMARK 5 2160 O2 in CO2 302
REMARK 5 2161 C in CO2 303
REMARK 5 2162 O1 in CO2 303
REMARK 5 2163 O2 in CO2 303
This means that PROPKA couldn’t process the ZN or CO2 residues.
Because of that, they weren’t included in the PQR, and thus were not in the
converted PDB.
Make a copy of the converted PDB to add the ZN and CO2 residues into.
cp 5Y2S_ph7_protonated.pdb 5Y2S_ph7_nsa.pdb
Now, you can copy the ZN and CO2 atom lines from 5Y2S_clean.pdb into
5Y2S_ph7_nsa.pdb.
As part of this:
- Change the temperature factor (the number after XYZ coordinates) to
0.00 - Remove the A segment ID
- Change
HETATMtoATOM
The 5Y2S_clean.pdb ZN line
HETATM 2157 ZN ZN A 301 14.442 0.173 15.262 1.00 3.61 ZN
becomes
ATOM 2157 ZN ZN 301 14.442 0.173 15.262 1.00 0.00 ZN
You can leave the atom ID (2157) as-is.
Further programs will renumber everything.
MolProbity
Upload the cleaned PDB file (5Y2S_ph7_nsa.pdb) with the corrected pH AMBER
types to MolProbity.
As part of the structure processing by MolProbity, all of the hydrogens are
removed.
These hydrogens must then be re-added with the “Add hydrogens” button.
Selecting this button brings up a new page with options.
Choose Asn/Gln/His flips as the method.
In this particular case, the 5Y2S structure comes from x-ray crystallography.
Thus, select Electron-cloud x-H for the bond length.
Then you can hit Start adding H >.
MolProbity found evidence to flip several residues.
Select the ones you’d like to flip with the check-boxes (all recommended)
before hitting Regenerate H, applying only selected flips >.
Because of the addition of hydrogens and flips, a pop-up appears.
Select yes to download the new structure now.
LEaP
LEaP works best with a PDB that’s as close to true form as possible. The new structure downloaded from MolProbity has some issues with it. We need to rectify these things:
TERlines should be between the protein and any metals, ions, or waters- Remove the
flipandnewmarks that MolProbity prints - Remove
USERandREMARKfrom PROPKA and MolProbity - Use atom names that are consistent with what LEAP expects
- In this case, change
OWforWATtoO
- In this case, change
This PDB file was cleaned up through by deleting the USER and REMARK heading
lines, as well as performing these string replacements with vi.
:%s/ flip//g
:%s/ new//g
:%s/ OW WAT/ O WAT/g
The other important thing we need to consider about this structure is its active site. The CO2 residue is not native to AMBER, so we need to get parameters for it. If this were a residue that had hydrogens (like a drug), then the anticipated hydrogens would need to be added before it was parametrized. Additionally, the ZN residue in the active site is one that would benefit from additional parameters. In this case, the ZN if 4-coordinate with 2 HID, 1 HIE, and 1 WAT. This is adequately described by ZAFF (which has its own tutorial).
Generating CO2 Parameters
Since CO2 is a small, organic molecule, it should be decently described by GAFF.
“Decently described” sounds sketchy…
Good! You picked up on that! It can be sketch. GAFF stands for General AMBER Force Field. That means it’s generalized for organic compounds that contain C, N, O, H, S, P, F, Cl, Br and I. So, if you have something that’s tricky, or that you’d want more tailored parameters for, you should use RESP fitting to compute them. One way to achieve this is to use pyRED.
To get the GAFF parameters, run antechamber.
Each of the flags gives the program information it needs to run.
| Flag | Purpose |
|---|---|
| -i | input file |
| -fi | file type of input file |
| -o | output file |
| -fo | file type of output file |
| -c | calculation type |
| -s | verbosity |
| -nc | net charge |
| -m | multiplicity |
antechamber -i CO2.pdb -fi pdb -o CO2.mol2 -fo mol2 -c bcc -s 2 -nc 0 -m 1
Ordinarily, after running antechamber, you run parmchk to make sure that
all of the necessary force field descriptors are present.
parmchk -i CO2.mol2 -f mol2 -o CO2.frcmod
This file contains no information, as everything required is covered by GAFF.
Incorporate ZAFF
ZAFF is the Zinc AMBER Force Field (read more). There are several parameter sets for for different 4-coordinate zinc complexes.
Looking at the 3D view on the RCSB website, you can identify that HID 94, HID 96, HIE 119, and WAT 455 are all coordinated to ZN 301 in the original structure. The 3 histidine, 1 water coordination is best described by ZAFF Center ID 4.
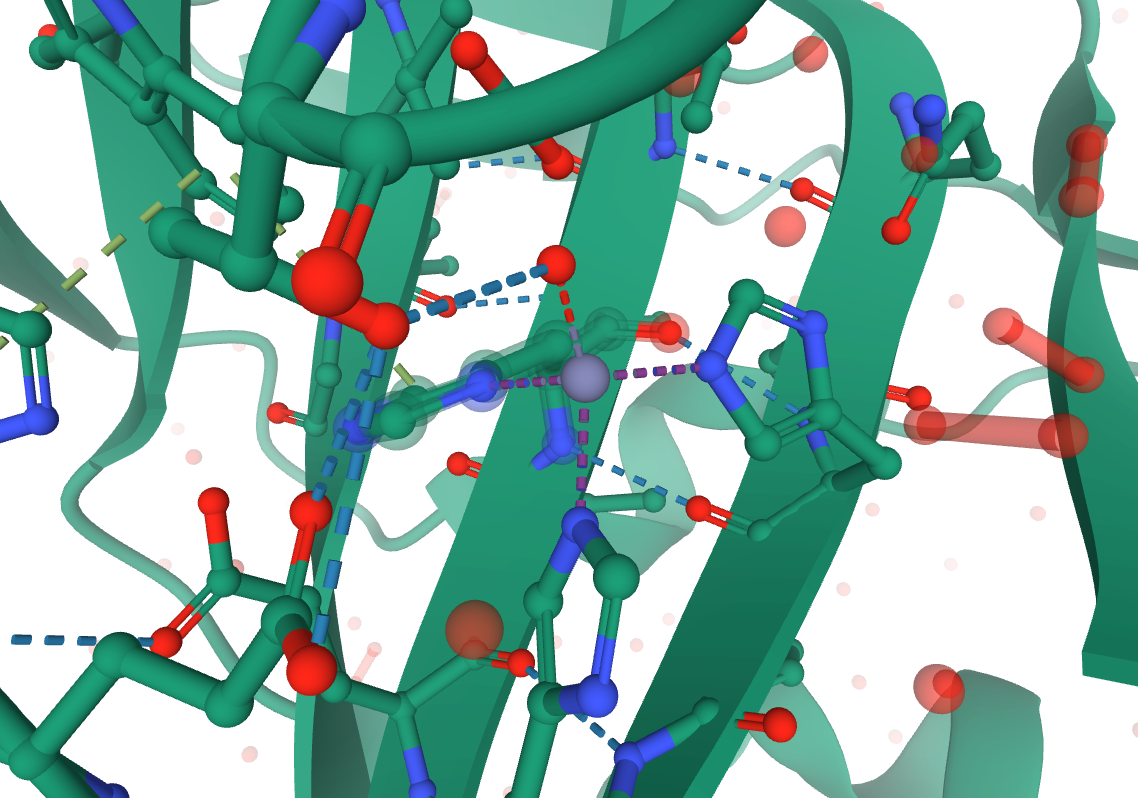
Image citation: 5Y2S, doi: 10.2312/molva.20181103, RCSB PDB.
Since PROPKA and MolProbity maintained the residue numbering, we can use those RESIDs to rename the coordinated residues as needed for ZAFF. Only the residue name (for the entire residue) needs to change.
| ResNum | Original ResName | New ResName |
|---|---|---|
| 94 | HID | HD4 |
| 96 | HID | HD5 |
| 119 | HIE | HE2 |
| 455 | WAT | WT1 |
| 301 | ZN | ZN6 |
Before moving on with LEaP, we need to download the parameter files for ZAFF. These can be downloaded from the ZAFF tutorial as a prep and frcmod. They are also included in the files downloaded for this lesson.
Using tleap
tleap is is the terminal form of LEaP.
There is also xleap, which uses a GUI.
Most of the time, people use an input script with tleap, which is evoked like:
tleap -f tleap.in
The tleap.in file for used here looks like:
source leaprc.gaff
source leaprc.protein.ff14SB
addAtomTypes { { "ZN" "Zn" "sp3" } { "N5" "N" "sp3" } { "N6" "N" "sp3" } { "N7" "N" "sp3" } }
source leaprc.water.tip3p
loadamberprep ZAFF.prep
loadamberparams ZAFF.frcmod
CO2 = loadmol2 CO2.mol2
c = loadpdb 5Y2S_ph7_mp_ZAFF.pdb
bond c.261.ZN c.94.NE2 ## HID NE2
bond c.261.ZN c.96.NE2 ## HID NE2
bond c.261.ZN c.119.ND2 ## HIE ND2
bond c.261.ZN c.318.O ## WAT O
savepdb c 5Y2S_vac.pdb
saveamberparm c 5Y2S_vac.prmtop 5Y2S_vac.inpcrd
addions c CL 0.0
solvatebox c TIP3PBOX 12.0
savepdb c 5Y2S_wat.pdb
saveamberparm c 5Y2S_wat.prmtop 5Y2S_wat_init0.rst
quit
First, the relevant force field information is sourced, and any new atom types
are defined.
Next, the information for ZAFF and CO2 are loaded in.
The CO2 = part corresponds to the 3-letter code that is in the PDB for that
residue.
Next, the protein is loaded in using a variable name, c.
Any variable name is fine, but if you update it, it’d need to be updated in
all of the remaining lines.
The bond lines generates a bond between the zinc and each of the atoms it
is coordinated to, which is an aspect of the ZAFF model.
The save lines save structures in various states.
There’s a first save before the ions and water are added, known as a “dry” or
“vacuum” structure.
Then, those ions (where 0.0 says to neutralize the charge) and waters are
added (where 12.0 specifies that water should extend 12.0 angstroms from the
protein’s surface).
These additions are saved in a “wet” or “solvated” structure.
LEaP does accept comments in these files, using #.
Note about
solvateboxMany people like to use
solvateoctto generate a truncated octahedron, which can save time by literally cutting corners, thus reducing the total number of solvent ions. If you are planning on performing QM/MM with LICHEM, it is very important to usesolvateboxbecause the truncated octahedron shape is not yet supported.
If you run this script on the provided files, it should work. However, if you were doing this on your own, you would likely encounter this error:
WARNING: The unperturbed charge of the unit: 0.998000 is not zero.
FATAL: Atom .R<NHIE 4>.A<HD1 20> does not have a type.
Failed to generate parameters
Parameter file was not saved.
Quit
The way to solve this error is to delete the HD1 hydrogen of the very first
residue, HIE 4, so that it can be rebuilt.
This is an error that can sometimes occur with first and last protein residues.
Testimonial from a Grad Student: Read the Error Messages
97.9% of my problems would be solved if I just read the error messages. Often, they tell you exactly what you need to do to fix it. If they don’t, I strongly recommend Google. Google is your best friend. Well, maybe not your best friend. I don’t even know you. I guess what I’m saying is that you’re probably not the first person to have an error of a specific type, which is why someone programmed an error message. Between the message, the internet, and the source code, there’s a lot of “clues” out there as to what’s going wrong.
Key Points
PROPKA is one way to calculate protonation states.
MolProbity can be used to check a structure for clashes, and make relevant ring flips.
Non-standard residues must have relevant hydrogens before parametrization.
For the final structure, LEAP will add hydrogens (and other missing sidechain atoms) based on residue definitions.
Molecular Dynamics and Clustering
Overview
Teaching: 60 min
Exercises: 60 minQuestions
How do I perform a molecular dynamics simulation with AMBER?
What are some of the different approaches to clustering?
How do I cluster a simulation based on specific criteria?
How do I do subclustering?
Objectives
Take an prmptop/inpcrd set through production.
Describe some benchmarks for determining whether a system is equilibrated.
Explain the difference between the k-means and DBscan clustering algorithms.
Highlights
- Molecular Dynamics Background
- Analyzing Molecular Dynamics Simulations
- Clustering
- Subclustering
- MPI Warning
- Writing a Frame for QM/MM
- MPI Warning
Molecular Dynamics Background
This where words would go, if I had them.
Analyzing Molecular Dynamics Simulations
This where words would go, if I had them.
Clustering
Clustering is a way to group together different snapshots of a trajectory. It can be done through random assignment, or using specific characteristics, such as a bond angle or distance.
cpptraj can be used to cluster trajectory data.
The k-means algorithm can be used to sort all trajectory points into a number
of defined clusters.
The DBscan algorithm can filter through outliers, but it requires a bit more
effort and testing to form clusters.
David Sheehan
has a great explanation of the different clustering algorithms.
trajin ../5Y2S_wat_imaged_1-50.nc
#trajin ../path/to/other/stripped/trajectories.nc
autoimage
## O-C Distance
distance d1 :259@O :261@C out 5Y2S_OC_dist.dat
## O-C-O Angle
## 2 kinds of OCO are possible!
angle a1 :259@O :261@C :261@O1 out 5Y2S_OCO1_ang.dat
angle a2 :259@O :261@C :261@O2 out 5Y2S_OCO2_ang.dat
## kdist test
cluster C0 dbscan kdist 9 data d1,a1,a2 sieve 10
## DBSCAN based on PA-O distance and OPO angle
cluster c1 dbscan minpoints 25 epsilon 2.2 data d1,a1,a2 \
pairdist 5Y2S_OC_OCO_db_clust_pairs.dat \
loadpairdist \
info 5Y2S_OC_OCO_db_clust_detail_info.dat \
out 5Y2S_OC_OCO_db_clustnum_v_time.dat \
summary 5Y2S_OC_OCO_db_clust_summary.dat \
avgout 5Y2S_OC_OCO_db_clust avgfmt pdb \
cpopvtime 5Y2S_OC_OCO_db_popvtime.dat
Because clustering is memory-intensive, we use stripped trajectories.
Typically, you will use whatever stripped trajectory you’ve created as part
of your analysis workflow.
In this case, we use the associated strip.5Y2S_wat_fix.prmtop file for the
topology information.
Since the overall goal of clustering is to sort frames into clusters based on critical criteria for the reaction, we can perform clustering on all of our replicates at once. Thus, you will generally use absolute paths to call all of the replicates one after another.
Subclustering
DBScan is is used to group frames into like categories, so there should be one or two clusters that arise with the proper characteristics for the proposed reaction. In order to more randomly select frames that match these criteria, you can recluster those within the matching cluster (what we refer to as subclustering).
To do this more easily, we use a grep command to separate out the list of
frames into their individual clusters.
$ grep " 0" 5Y2S_OC_OCO_db_clustnum_v_time.dat > clust_num_0.txt
$ grep " 1" 5Y2S_OC_OCO_db_clustnum_v_time.dat > clust_num_1.txt
$ grep " 2" 5Y2S_OC_OCO_db_clustnum_v_time.dat > clust_num_2.txt
...
From the clust_num_X.txt file with the closest match, you can use Python to
print a list of frames to then use in your subclust.in file with cpptraj.
You don’t have to use Python, it’s just one way we’ve chosen to highlight.
import pandas as pd
clust = pd.read_csv("clust_num_0.txt", header=None, delim_whitespace=True)
test = clust.loc[:,0].values.tolist()
test2 = ''.join(str(i)+"," for i in test)
f=open("out_clust0.txt","w+")
f.write(test2)
f.close()
You can then write a subclust.in file that writes out the relevant frames
to a new trajectory file, and cluster those frames separately.
Use the strip.5Y2S_wat_fix.prmtop file for the topology information.
trajin ../../5Y2S_wat_imaged_1-50.nc
autoimage
## Center the non-solvent residues -- VERY IMPORTANT FOR TINKER
center :1-261 origin mass
## Write out the frames of a single cluster, as identified through the
## `grep` command
trajout 5Y2S_wat_subclust_num_0.nc netcdf onlyframes \
232,524,1041,1259,1788,1841,1847,1856,1869,1873,2280,2293,2600,2663,2710,\
2716,2717,2877,2977,2999,3130,3343,3360,3374,3606,3616,3618,3629,5036,5044,\
5240,5244,5415,5427,5473,5998,6168,6334,6704,6750,7056,7863,7899,8685,8756,\
8810,9344,9802,9911,9916,9941,9980,10747,10756,10804,10806,10818
autoimage
## Use this next block to create that file and clear any currently loaded
## files
#####################
go
clear trajin
go
####################
## Read in the file with specific frames from the cluster from the file you
## just wrote
trajin 5Y2S_wat_subclust_num_0.nc
## O-C Distance
distance d1 :259@O :261@C
## O-C-O Angle
## 2 kinds of OCO are possible!
angle a1 :259@O :261@C :261@O1
angle a2 :259@O :261@C :261@O2
## ## 10 clusters of k-means based on OC distance and OCO angles
cluster coco kmeans clusters 10 data d1,a1,a2 \
info 5Y2S_OC_OCO_sub_km_clust_detail_info.dat \
out 5Y2S_OC_OCO_sub_km_clustnum_v_time.dat \
summary 5Y2S_OC_OCO_sub_km_clust_summary.dat \
avgout 5Y2S_OC_OCO_sub_km_clust avgfmt pdb \
cpopvtime 5Y2S_OC_OCO_sub_popvtime.dat
MPI Warning
When you go to run this subclustering, DO NOT use the MPI version of cpptraj! It has issues with reading and writing individual frames!
One advantage to subclustering is that you can use different criteria for the first and second clustering passes. For instance, if you have an angle and a distance that are likely important for the first reaction step, you can do the inital clustering on that and the subclustering using the angle and distance of a secondary reaction step.
Writing a Frame for QM/MM
After performing subclustering, you can once again use cpptraj to write PDBs
of the selected frames.
These frames are the snapshots that will be used for QM/MM.
However, it’s not straightfoward, because you perform clustering on stripped
trajectories, and you need unstripped trajectories for QM/MM.
Thus, we’ll use a script similar to how we did subclustering in order to write the frames we need.
First, load in all of the frames you used when you created the unstripped trajectory.
After subclustering, you can use the 5Y2S_OC_OCO_sub_km_clust_summary.dat
file to write the centroids of the 10 clusters identified through k-means.
If you rerun the analysis, these centroids should be different, as k-means
is random!
Use the 5Y2S_wat_fix.prmtop file for the topology information.
## Read in all the files used for to create the stripped trajectories in the
## exact same order to ensure that the proper frames are pulled
trajin /absolute/path/to/the/file/for/replicate1/5Y2S_wat_md1.mdcrd
trajin /absolute/path/to/the/file/for/replicate1/5Y2S_wat_md2.mdcrd
trajin /absolute/path/to/the/file/for/replicate2/5Y2S_wat_md1.mdcrd
trajin /absolute/path/to/the/file/for/replicate2/5Y2S_wat_md2.mdcrd
trajin /absolute/path/to/the/file/for/replicate3/5Y2S_wat_md1.mdcrd
trajin /absolute/path/to/the/file/for/replicate3/5Y2S_wat_md2.mdcrd
## ... continue
autoimage
## Center the non-solvent residues -- VERY IMPORTANT FOR TINKER
center :1-261 origin mass
## Write out the frames of a single cluster, as identified through the
## `grep` command
trajout 5Y2S_wat_subclust_num_0_unstripped.nc netcdf onlyframes \
232,524,1041,1259,1788,1841,1847,1856,1869,1873,2280,2293,2600,2663,2710,\
2716,2717,2877,2977,2999,3130,3343,3360,3374,3606,3616,3618,3629,5036,5044,\
5240,5244,5415,5427,5473,5998,6168,6334,6704,6750,7056,7863,7899,8685,8756,\
8810,9344,9802,9911,9916,9941,9980,10747,10756,10804,10806,10818
## Use this next block to create that file and clear any currently loaded
## files
#####################
go
clear trajin
go
####################
## Read in the file with specific frames from the cluster from the file you
## just wrote
trajin 5Y2S_wat_subclust_num_0_unstripped.nc
autoimage
## Center the non-solvent residues -- VERY IMPORTANT FOR TINKER
center :1-261 origin mass
## Write out the specific PDBs identified with clustering
trajout 5Y2S_subclust_c0_frame_6.pdb pdb onlyframes 6
trajout 5Y2S_subclust_c1_frame_29.pdb pdb onlyframes 29
trajout 5Y2S_subclust_c2_frame_34.pdb pdb onlyframes 34
trajout 5Y2S_subclust_c3_frame_56.pdb pdb onlyframes 56
trajout 5Y2S_subclust_c4_frame_10.pdb pdb onlyframes 10
trajout 5Y2S_subclust_c5_frame_45.pdb pdb onlyframes 45
trajout 5Y2S_subclust_c6_frame_4.pdb pdb onlyframes 4
trajout 5Y2S_subclust_c7_frame_5.pdb pdb onlyframes 5
trajout 5Y2S_subclust_c8_frame_47.pdb pdb onlyframes 47
trajout 5Y2S_subclust_c9_frame_5.pdb pdb onlyframes 5
MPI Warning
When you run this input, DO NOT use the MPI version of cpptraj! It does not support writing individual frames!
Start by reading in the file with the subclusters, make sure that they are all
autoimaged and recentered, and then write them out with the onlyframes
argument.
Key Points
An MD simulation can be broken down into 4 phases: minimization, heating, equilibration, and production.
DBscan can be very finicky, but it is a better approach to clustering.
Run k-means after DBscan. You can select the centroids from k-means as the random snapshots.
QM/MM Structure Preparation
Overview
Teaching: 60 min
Exercises: 60 minQuestions
How is a Tinker XYZ file different from a regular XYZ file?
How do I convert a PDB file to a Tinker XYZ file?
How do I write a Gaussian BASIS file?
How do I use LICHEM for a single point energy calculation?
Objectives
Learn about the different files required for a LICHEM calculation.
Modify a Python script with the proper QM region to generate the
regions.inpfile.Submit your first single point energy calculation with LICHEM.
Highlights
- Overview of the QM/MM Process
- The Trials
and Tribulationsof PDB to XYZ - Generating the TINKER Parameter File
- Converting the PDB
- Potential Script Issue
- Checking the XYZ with
analyze - Building
regions.inpand Converting to LICHEM - Selected QM/MM Citations for Human Carbonic Anhydrase
- Get Your Numbers Straight
- Running a Single Point Energy Calculation
- Version Note
Overview of the QM/MM Process
- Set-up and run MD
- Cluster the trajectory (see phase 3)
- Write out an unstripped frame centered on the origin
- Build a parameter file!
- Convert to TINKER XYZ
- Build the
regions.inp,connect.inp,tinker.key, andBASISfile.- Run an SP
- Do a DFP for the reactant (phase 5)
- Build the product from the optimized reactant (phase 5)
- Compare the product and optimized reactant (phase 5)
- QSM (phase 6)
The Trials and Tribulations of PDB to XYZ
TINKER uses a specific type of XYZ file, aptly referred to as a TINKER XYZ. The first line contains the total number of atoms in the file (and sometimes a comment). Any following lines start with the atom number, the atom name, the XYZ coordinates, the Tinker atom type, and any atoms that that atom is connected to.
43380
1 N 22.109000 -3.927000 -6.090000 1 2 3 4 5
2 H1 22.156000 -4.914000 -5.880000 2 1
You need to start with a Tinker XYZ file to build the relevant LICHEM XYZ and connect files. These two files are read into LICHEM (with some other information), and LICHEM will then handle any inter-program file conversions.
If you’re using AMOEBA as a force field after running AMBER MD, you will need to have a set of AMOEBA parameters for any non-standard residues. Since this tutorial involves the non-standard residue CO2, and we do not have AMOEBA parameters for it, we will be using the point charge method for QM/MM.
You can use Tinker’s pdbxyz program, but the program tends to crash
anytime something goes wrong, leaving you without any part of the new
Tinker XYZ, even if there was only one bad atom.
For this reason, several of us have coded ways around this program.
Here, we will use the
generate_TINKER_parameters.py
script in conjuction with the
pdbxyz4amber-pmd-params.py.
This script will use the original prmtop file to pull the same paramters that
were used for the AMBER MD and write them into a custom Tinker parameter file.
Because it takes the existing parameters, the non-standard residues are covered.
I have a few other means for the conversion and back-conversion of PDBs and
Tinker XYZs in my
pdbxyz-xyzpdb repository.
Mark has also written a Python package,
PDBTinker to convert PDB files
to a Tinker XYZ using an AMOEBA force field.
Generating the TINKER Parameter File
As stated, since we’re using AMBER for the QM/MM, we will be using the
generate_TINKER_parameters.py
script to build our parameter file.
This parameter file is independent of the TINKER version used.
Modify the options above the Definitions line, in this case source_params
and param_file_name.
## !!! This uses Pandas 1.0 !!! Without it, remove 'ignore_index' in the
## drop_duplicates lines.
import parmed as pmd
import pandas as pd
import numpy as np
import copy
from collections import OrderedDict
## Code to source a single parm file (not a leaprc)
# source_params = "parm99.dat"
# param_dat = pmd.load_file(source_params)
## Prmtop Method
source_params = "5Y2S_wat_fix.prmtop"
param_dat = pmd.load_file(source_params)
## It looks like anything sourced in the leaprc needs to have an absolute
## path to it, so you might need to modify the leaprc to incorporate the
## absolute path. It is STRONGLY RECOMMENDED that you copy the leaprc file to
## do this, and then reference that copy!!
## Leaprc Method
# source_params = "param_files/leaprc.ff14SB.OL15.tip3p"
# param_dat = pmd.amber.AmberParameterSet().from_leaprc(source_params)
## Leave the X dihedrals as X to fix by hand
## Setting this false will likely result in a MAXPRM issue with TINKER
## Because... well... you'll likely have over a million dihedral angles.
leave_as_X = True
## Give your FF a name (it will be preceded by AMBER-)
ff_name = "ff14SB"
## Give your new parameter file a name
param_file_name = "5Y2S_TINKER_params.prm"
Then run the script.
$ python3 generate_TINKER_parameters.py
Converting the PDB
Now that we have a functional parameter file, we can convert our PDB using
pdbxyz4amber-pmd-params.py.
This script takes the cluster PDB file (infile) and the paramater file
(param_file) as input.
Like before, we will modify the first few lines with the file names.
import parmed as pmd
import numpy as np
import pandas as pd
import re
import sys
infile="5Y2S_subclust_c0_frame_6.pdb"
outfile="5Y2S_subclust_c0_frame_6.xyz"
param_file="5Y2S_TINKER_params.prm"
atom_lines="atom-lines.txt"
test_csv="test.csv"
Then run the script.
$ python3 pdbxyz4amber-pmd-params.py
This script will usually take about 3 minutes to run because it consists of a bunch of if statements that each atom has to go through.
Potential Script Issue
As Python updates, so do certain errors. It appears that
UnboundLocalErrormay not be how Pandas refers to match errors now, instead usingNameError. If you get:Traceback (most recent call last): File "pdbxyz4amber-pmd-params.py", line 256, in <module> system = convert_names(system, lines, AMOEBA) File "pdbxyz4amber-pmd-params.py", line 177, in convert_names print(residue.name, atom.name, test_RN, test_name) NameError: name 'test_RN' is not definedChange
except UnboundLocalError:toexcept NameError:on line 177.
When the script runs properly, it should print:
Processing as AMBER parameters.
If I didn't find residues, they'll be listed here:
Residue Name | Atom Name | Search ResName | Search Atom Name
ZN ZN ZN ZN
This alerts you that ZN is not being selected properly based on either your
PDB file or the parameter file.
A quick check of our PDB file shows that the ZN atom name is offset by a
column, but this doesn’t actually affect the script.
ATOM 4054 OXT LYS 257 -20.102 -10.326 14.057 1.00 0.00 O
ATOM 4055 ZN ZN6 258 3.179 -0.916 -1.326 1.00 0.00 ZN
ATOM 4056 H1 WT1 259 5.491 0.309 -0.943 1.00 0.00 H
Our parameter file also doesn’t look particularly alarming.
atom 435 38 ZN "ZN6 ZN" 30 65.41 0 !! GUESSED CONNECTION
For some reason, it just doesn’t like the ZN6, likely due to an internal
attempt at removing the final number to search for matches.
If we wanted to, we could build an exception in for when we run this script
in the future.
This is the easiest option if you’re going to be testing more than one
snapshot, since you can just keep reusing the script.
Uncomment the first batch of lines under ## Address problem residues! and
modify them accordingly.
## Address problem residues!
if atom_test.empty == True:
if residue.name in ('ZN'): ## We know it's ZN because of the print-out
test_RN = 'ZN6' ## It needs to be looking for ZN6 in the PDB
test_name = 'ZN' ## ZN6 has an atom name of ZN
res_test = lines[lines.ResName == test_RN]
atom_test = res_test[res_test.AtomName == test_name]
If we don’t want to modify the Python script, we can directly add the type to the Tinker XYZ. In this approach, we change:
4055 ZN 3.179000 -0.916000 -1.326000 0 ATOM TYPE NOT FOUND
to
4055 ZN 3.179000 -0.916000 -1.326000 435
in the Tinker XYZ that the script created.
Checking the XYZ with analyze
After generating the Tinker XYZ, it is absolutely crucial to verify that
there are no missing parameters for TINKER.
Parameter issues cause major headaches, and it’s so much easier to run analyze
than try and debug a QM/MM calculation.
You must use the analyze corresponding to whatever version of TINKER you
intend on using, as it changes across versions.
Before running analyze, however, we must create a tinker.key file.
The tinker.key file tells TINKER what parameter file it needs to use.
This key file only needs to contain one line (for now).
parameters 5Y2S_TINKER_params.prm
After you’ve created the tinker.key, you can run analyze.
$ analyze 5Y2S_subclust_c0_frame_6.xyz
In our case, we’re missing parameters! Oh, joy! Because they’re water parameters, it prints a gazillion and you can’t see what the actual issue is. So, you can modify your analyze command, or save it to an output file.
$ analyze 5Y2S_subclust_c0_frame_6.xyz | head -n 50
Now we can determine what it doesn’t like!
Atoms with an Unusual Number of Attached Atoms :
Type Atom Name Atom Type Expected Found
Valence 1417-N5 345 3 2
Valence 1454-N6 362 3 2
Valence 1797-N7 375 3 2
Valence 4059-c1 439 0 2
Valence 4060-o 440 0 1
Valence 4061-o 441 0 1
Valence 4062-c1 439 0 2
Valence 4063-o 440 0 1
Valence 4064-o 441 0 1
Undefined Angle Bending Parameters :
Type Atom Names Atom Classes
Angle 4067-HW 4066-OW 4068-HW 39 44 39
Angle 4070-HW 4069-OW 4071-HW 39 44 39
Angle 4073-HW 4072-OW 4074-HW 39 44 39
It’s upset about the connectivity of several atoms (those in residues
HD4, HD5, HE2, and CO2, which makes sense because they’re not typical
protein residues).
The 3 atoms listed as part of HD4, HD5, and HE2 are all the nitrogen
coordinated to the zinc atom.
Since the zinc coordination itself is not considered a bond, we can decrease
the expected number of connections in the parameter file.
atom 345 32 N5 "HD4 NE2" 7 14.01 2 !! GUESSED CONNECTION
atom 362 33 N6 "HD5 NE2" 7 14.01 2 !! GUESSED CONNECTION
atom 375 34 N7 "HE2 ND1" 7 14.01 2 !! GUESSED CONNECTION
[Note: you can remove the !! GUESSED CONNECTION if you want, since it’s just
a comment anyway.]
The inferred CO2 coordination is not correct, so its expected number of
connections also need to be adjusted in the parameter file.
atom 439 41 c1 "CO2 C" 6 12.01 2 !! GUESSED CONNECTION
atom 440 42 o "CO2 O1" 8 16.00 1 !! GUESSED CONNECTION
atom 441 42 o "CO2 O2" 8 16.00 1 !! GUESSED CONNECTION
The undefined water parameters also make sense, as stated in the README for
the generate_TINKER_parameters.py script.
Building the parameter file drops the angle information from the solvent
(angle HW OW HW 100.00 104.52), so we have to append it to the angle section.
angle 39 44 39 100.0 104.52
After modifying the parameter file with these changes, we can re-run analyze.
Enter the Desired Analysis Types [G,P,E,A,L,D,M,V,C] : E
Total Potential Energy : -123334.5645 Kcal/mole
Intermolecular Energy : -118769.0709 Kcal/mole
Energy Component Breakdown : Kcal/mole Interactions
Bond Stretching 795.9634 30331
Angle Bending 2286.9420 20568
Improper Torsion 589.9997 4968
Torsional Angle 0.0000 11012
Van der Waals 16585.5434 146715508
Charge-Charge -143593.0130 940839611
Great! No problems this time, and a nice, negative Charge-Charge energy.
Huzzah!!!!!!
Now go back to that fixed parameter file and remove the !! GUESSED CONNECTION
comments (a string replacement, like %s/!! GUESSED CONNECTION//g will work fine).
These end-of-line comments can cause a segmentation fault when doing file
conversion in LICHEM.
Building regions.inp and Converting to LICHEM
Now that we have a Tinker XYZ, we can make use the
create_reg.py script
to create our regions.inp file.
This script is a skeleton and will be active-site specific, so we need to figure out some things first! [Insert reference to selecting QM region]
Our QM region for 5Y2S will include the ZN6, HD4, HD5, HE2, WT1,
CO2, any waters within a specific cutoff of the ZN6 and CO2, and
HIS 64 in our active site.
In several QM/MM papers, HIS 64 (the biologically relevant name for what is
actually residue 61 in our PDB file) is important for the reaction.
Selected QM/MM Citations for Human Carbonic Anhydrase
- Zheng, Y. J. and Merz Jr., K. M. J. Am. Chem. Soc. 1992, 114, 26, 10498–10507. DOI: 10.1021/ja00052a054
- Chen, H.; Li, S.; Jiang, Y. J. Phys. Chem. A 2003, 107, 23, 4652–4660. DOI: 10.1021/jp026788k
- Riccardi, D. and Cui, Q. J. Phys. Chem. A 2007, 111, 26, 5703–5711. DOI: doi.org/10.1021/jp070699w
Now that we know what to include in our active site, we can modify the
create_reg.py script.
First, we modify the header information, selecting the zinc residue as the center of our shell around the QM region.
orig_pdb="5Y2S_subclust_c0_frame_6.pdb"
tink_xyz="5Y2S_subclust_c0_frame_6.xyz"
## Atom number for center of active atom shell
shell_center=4055
## Did you use the index from VMD for the shell_center? If yes, set True
VMD_index_shell=False
After changing the header, we have to re-write the
select_QM(system, shell_center) function.
You can use either VMD numbering (which starts are zero) or Tinker XYZ/PDB
numbering (which start at 1), but you must use the proper syntax for each.
It is incredibly easy to get the numbers wrong.
The table below contains the assignments for the QM atoms to use in the script.
In this structure, HN refers to H, but is shown as HN because
other preparation programs name the H on the backbone nitrogen accordingly.
It uses the atom numbers within the Tinker XYZ/PDB.
| QM Residue(s) | Number | Pseudobond (CA, CB) | Boundary | Charge | |
|---|---|---|---|---|---|
| XYZ/PDB number | XYZ/PDB number | XYZ/PDB number | |||
| HID 61 (940 - 956) | 944 - 954 | 11 | CA 942 | C (955), HA (943), N (940), O (956), HN (941) | 0 |
| HD4 (1405 - 1421) | 1409 - 1419 | 11 | CA 1407 | C (1420), HA (1408), N (1405), O (1421), HN (1406) | 0 |
| HD5 (1442 - 1458) | 1446 - 1456 | 11 | CA 1444 | C (1457), HA (1445), N (1442), O (1458), HN (1443) | 0 |
| GLU 103 (1574 - 1588) | 1581 - 1586 | 6 | CB 1578 | C (1587), HA (1577), N (1574), O (1588), HN (1575), CA (1576), HB2 (1579), HB3 (1580) | 1- |
| HE2 (1789 - 1805) | 1793 - 1803 | 11 | CA 1791 | C (1804), HA (1792), N (1789), O (1805), HN (1790) | 0 |
| ZN6 (4055) | 4055 | 1 | 2+ | ||
| WT1 (4056 - 4058) | 4056 - 4058 | 3 | - | - | 0 |
| CO2 (4062 - 4064) | 4062 - 4064 | 3 | - | - | 0 |
| WAT | ??? | WAT | 0 | ||
| Total: | 57 + WAT | 5 | 28 | 1+ |
Once we know what our QM, pseudobond, and boundary atoms are, we can modify that function.
def select_QM(system, shell_center):
## My QM Atoms
QM_HID_61 = system.select_atoms("bynum 944:954")
QM_HD4_91 = system.select_atoms("bynum 1409:1419")
QM_HD5_93 = system.select_atoms("bynum 1446:1456")
QM_GLU_103 = system.select_atoms("bynum 1581:1586")
QM_HE2_116 = system.select_atoms("bynum 1793:1803")
QM_ZN6_258 = system.select_atoms("resnum 258")
QM_WT1_259 = system.select_atoms("resnum 259")
QM_CO2_261 = system.select_atoms("bynum 4062:4064")
QM_WAT = system.select_atoms("(around 4 resnum 258) or (around 4 resnum 61) and (resname WAT)")
QM_WAT = QM_WAT.residues.atoms
#
## Combine the QM atoms. Consider using `|` instead of '+' to make `all_QM`
## ordered with a single copy of an atom.
all_QM = QM_HID_61 | QM_HD4_91 | QM_HD5_93 | QM_GLU_103 | QM_HE2_116 | QM_ZN6_258 | \
QM_WT1_259 | QM_CO2_261 | QM_WAT
#
## My Pseudo Atoms
PB_HID_61 = system.select_atoms("resnum 61 and name CA")
PB_HD4_91 = system.select_atoms("resnum 91 and name CA")
PB_HD5_93 = system.select_atoms("resnum 93 and name CA")
PB_GLU_103 = system.select_atoms("resnum 103 and name CB")
PB_HE2_116 = system.select_atoms("resnum 116 and name CA")
#
## Combine the PB atoms. Consider using `|` instead of '+' to make `all_PB`
## ordered with a single copy of an atom.
all_PB = PB_HID_61 | PB_HD4_91 | PB_HD5_93 | PB_GLU_103 | PB_HE2_116
#
## My Boundary Atoms
BA_HID_61 = system.select_atoms("resnum 61 and (name N or name H or name HA \
or name C or name O)")
BA_HD4_91 = system.select_atoms("resnum 91 and (name N or name H or name HA \
or name C or name O)")
BA_HD5_93 = system.select_atoms("resnum 93 and (name N or name H or name HA \
or name C or name O)")
BA_GLU_103 = system.select_atoms("resnum 103 and (name N or name H or name HA \
or name C or name O or name CA or name HB2 or name HB3)")
BA_HE2_116 = system.select_atoms("resnum 116 and (name N or name H or name HA \
or name C or name O)")
#
## Combine the BA atoms. Consider using `|` instead of '+' to make `all_BA`
## ordered with a single copy of an atom.
all_BA = BA_HID_61 | BA_HD4_91 | BA_HD5_93 | BA_GLU_103 | BA_HE2_116
#
## Combine the BA atoms. Consider using `|` instead of '+' to make `all_BA`
## ordered with a single copy of an atom.
all_BA = BA_HID_61 | BA_HD4_91 | BA_HD5_93 | BA_GLU_103 | BA_HE2_116
#
print("There are {} QM, {} pseudobond, and {} boundary atoms.\n".format( \
len(all_QM), len(all_PB), len(all_BA)))
#
...
Another area we likely want to modify is the make_regions(...) function.
That function is what is used to write out the regions.inp_backup file, and
has some presets in there, such as the amount of memory used (QM_memory),
QM_charge and QM_spin.
If you’re only setting up this single frame, it’s alright if you leave it
as the preset in the script, as long as you modify the regions.inp file
before you use it for your calculation.
If you’re setting up multiple frames, typically you’ll figure this out for one
frame, modify the script, and then copy it for any remaining snapshots that
need to be prepared.
After modification, you can run the script.
$ python3 create_reg.py
This script will create files named regions.inp_backup and BASIS_list.txt.
The reason for naming the regions.inp file with _backup is because running
the LICHEM conversion will replace any existing files named connect.inp,
regions.inp, or xyzfile.xyz in the current directory with the newly
generated one.
In the case of regions.inp, this new copy would only contain
QM_atoms: 0
Pseudobond_atoms: 0
Boundary_atoms: 0
Frozen_atoms: 0
getting rid of all the hard work you went through to create it!
To actually convert the Tinker XYZ, we use:
$ lichem -convert -t 5Y2S_subclust_c0_frame_6.xyz -k tinker.key > conversion-1.log
Because we’re using Gaussian, we need a BASIS file.
We can use another LICHEM command to create a skeleton BASIS file.
However, depending on the version of LICHEM that you’re using, g16 may
not be recognized as a keyword.
So, copy the regions file we made using create_reg.py
cp regions.inp_backup regions.inp_BASIS
and change the QM_type to Gaussian.
This should be semi-commented out below it, but the syntax for a comment cannot
have spaces, whereas keyword searches do need a space after the colon.
Thus, the toggled version would resemble:
!QM_type:g16
QM_type: Gaussian
You can then create the BASIS file using this file.
$ lichem -convert -b regions.inp_BASIS
For the BASIS file, the numbers are based on the numerical order of what is
listed in QM and pseudobonds sections of regions.inp.
This means that if you have numbers 1123 1433 1353 listed in different places
in the file, 1123 = 1, 1353 = 2, and 1433 = 3.
Get Your Numbers Straight
- VMD: Starts atom numbering at zero (0).
- LICHEM: Starts atom numbering at zero (0).
- BASIS: Starts at one (1), but counts using only the atoms identified in the QM, PB, and BA sections of the
regions.inpfile.- TINKER: Starts atom numbering at one (1).
- AMBER: Starts atom numbering at one (1).
To make modifying this file easier, BASIS_list.txt maps the atoms between
VMD numbering, PDB numbering, and the Gaussian BASIS numbering.
All of this helps us ensure that the atoms we want to treat with a higher
basis, or that need have the pseudopotential atoms applied, are correct!
As-is, the last few lines of this file look like:
78 79 80 81 82 83 84 85 0
GEN
****
86 87 88 89 90 91 92 93 0
GEN
****
7 19 31 43 50 0
[PB basis set]
****
7 19 31 43 50 0
[PB pseudopotentials]
Throughout the file, GEN is used to mean “General BASIS”.
For this tutorial, we will be using 6-31G* for our QM region and 6-31+G(d,p)
for any reactive atoms in the QM region.
LICHEM can’t guess what our reactive atoms are, so we need to pull them out
from the numeric list that it sorts atoms into and specify them separately.
Formatting is very important in this file, and follows the pattern of
any relevant numbers, two spaces, a zero, followed by a newline with the basis
set, followed by a newline with four asterisks.
#1 #2 #3__0
GEN
****
Here, we want to make sure the atoms from residue CO2 and WT1, as well as the metal, are treated with a higher basis. Luckily for us, these are all sequential, so we need to select 56-62.
54 55 56 0
6-31G*
****
63 64 65 66 67 68 69 0
6-31G*
****
57 58 59 60 61 62 0
6-31+G(d,p)
****
It doesn’t matter if they’re in sequential order in the file–what matters is that each atom is only listed once.
The pseudopotentials for the pseudobond atoms also need to be modified.
1 13 25 37 44 0 STO-2G
SP 2 1.00
0.9034 1.00 1.00
0.21310 1.90904 0.57864
****
1 13 25 37 44 0
try1 1 2
S Component
1
1 7.75 16.49
P
1
1 1.0 0.0
After this, you should be ready for a single point energy calculation. You’re so close!
Running a Single Point Energy Calculation
Before you run a single point calculation, you should create a shell file to
run the LICHEM job (or some sort of queue script).
In the below example, 20 processors were used to run LICHEM, and a variable
named tag was created to name the output files.
tag=5Y2S_subclust_c0_frame_6_out
## -n number of processors
## -x input xyz
## -c connectivity file
## -r regions file
## -o output xyz file
## -l output log file
lichem -n 20 -x xyzfile.xyz -c connect.inp -r regions.inp -o ${tag}.xyz -l ${tag}.log
After that script is made, verify that you have the following files in the run directory:
5Y2S_TINKER_params.prmBASISconnect.inpregions.inprun-serial.sh(or whatever the run script is named)tinker.keyxyzfile.xyz
You can then submit that job through whatever means you are accustomed to.
Once it starts, you should check it to make sure it’s running properly.
You can do this through watch tail 5Y2S_subclust_c0_frame_6_out.log.
If you have a positive energy at any point, kill the job.
- If it’s in the QM region, something went wrong with the QM portion–debug that.
- If it’s in the MM region, something went wrong with the MM portion–debug that.
- If it’s in both, something catastrophic happened and everything failed–start debugging in the QM.
If everything worked properly, you should get this in your
5Y2S_subclust_c0_frame_6_out.log file:
Single-point energy:
QM energy: -4335.8971333871 a.u.
MM energy: -209.73460474142 a.u.
QMMM energy: -4545.6317381286 a.u. | -2852426.9841024 kcal
Version Note
An older version of LICHEM would look like:
Single-point energy: QM energy: -117985.76643379 eV MM energy: -5707.1690879283 eV QMMM energy: -123692.93552171 eV -4545.6317381286 a.u.
It’s party time! You did your first single point energy calculation!!! Seriously, it took a lot of work to get here, and you should celebrate. Go get yourself a calming beverage and come back to the tutorial.
Key Points
Parameters are, and always be, the downfall of the computational chemist.
File numbering is a really common trouble spot, so pay close attention to what you’re working with and where.
DFP Optimizations and Strategies for Building a Suitable Product
Overview
Teaching: 60 min
Exercises: 0 minQuestions
???
Objectives
???
Highlights
- Cluster the trajectory (see phase 3)
- Write out an unstripped frame centered on the origin
- Build a parameter file!
- Convert to TINKER XYZ
- Build the
regions.inp,connect.inp,tinker.key, andBASISfile. - Run an SP
- Do a DFP for the reactant (phase 5)
- Build the product from the optimized reactant (phase 5)
- Compare the product and optimized reactant (phase 5)
- QSM (phase 6)
Key Points
???
QSM: The Quantum String Method
Overview
Teaching: 60 min
Exercises: 0 minQuestions
???
Objectives
???
Highlights
- Cluster the trajectory (see phase 3)
- Write out an unstripped frame centered on the origin
- Build a parameter file!
- Convert to TINKER XYZ
- Build the
regions.inp,connect.inp,tinker.key, andBASISfile. - Run an SP
- Do a DFP for the reactant (phase 5)
- Build the product from the optimized reactant (phase 5)
- Compare the product and optimized reactant (phase 5)
- QSM (phase 6)
Key Points
???